
Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents
by
 , , and
, , and
Paula Teixeira
1,* ,
,
Carmen Bacariza
1 ,
,
Patrícia Correia
1, 
Carla I. C. Pinheiro
1 and
and
Isabel Cabrita
2,3,*
1
Centro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal
2
Institute of Higher Studies on Education and Sciences, ISEC Lisboa, School of Management, Engineering and Aeronautics, 1750-142 Lisboa, Portugal
3
CERENA, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal
*
Authors to whom correspondence should be addressed.
Energies 2022, 15(11), 4039; https://doi.org/10.3390/en15114039
Submission received: 26 April 2022
/
Revised: 18 May 2022
/
Accepted: 23 May 2022
/
Published: 31 May 2022
(This article belongs to the Special Issue Advances in Hydrogen Energy Production and Storage)
Abstract
:Hydrogen is a versatile vector for heat and power, mobility, and stationary applications. Steam methane reforming and coal gasification have been, until now, the main technologies for H2 production, and in the shorter term may remain due to the current costs of green H2. To minimize the carbon footprint of these technologies, the capture of CO2 emitted is a priority. The in situ capture of CO2 during the reforming and gasification processes, or even during the syngas upgrade by water–gas shift (WGS) reaction, is especially profitable since it contributes to an additional production of H2. This includes biomass gasification processes, where CO2 capture can also contribute to negative emissions. In the sorption-enhanced processes, the WGS reaction and the CO2 capture occur simultaneously, the selection of suitable CO2 sorbents, i.e., with high activity and stability, being a crucial aspect for their success. This review identifies and describes the solid sorbents with more potential for in situ CO2 capture at high and medium temperatures, i.e., Ca- or alkali-based sorbents, and Mg-based sorbents, respectively. The effects of temperature, steam and pressure on sorbents’ performance and H2 production during the sorption-enhanced processes are discussed, as well as the influence of catalyst–sorbent arrangement, i.e., hybrid/mixed or sequential configuration.
1. Introduction
The European Green Deal [1] has the overarching aim of making Europe climate neutral in 2050, but global energy consumption is expected to continue growing, requiring decarbonized energy vectors for end use applications [2]. Hydrogen is a promising energy vector, with a high calorific value (122 kJ g−1), which is being considered as the cleanest energy option, with a zero-carbon footprint, since it burns cleanly, giving water as the only product. Therefore, if linked with renewable energy sources and CO2 capture, it allows for decarbonizing a wide range of final sectors of use, providing clean power and heat to transport and stationary applications [3]. Hydrogen is an important raw material of some industrial processes, such as hydrocracking, ammonia synthesis, methanol production, and the manufacture of hydrochloric acid; it is also a reducing agent in the steel industry [4]. Up to now, 96% of the H2 production technologies [5] are based on non-renewable sources such as the steam methane reforming (SMR) of natural gas/oil-based or coal gasification (grey H2). The H2 production followed by CO2 capture is an interesting alternative to reduce carbon footprint but is in an early stage of implementation (blue H2).
The transition to a more sustainable H2 production cannot be dissociated from economic and technological aspects. Water electrolysis is a mature technology that produces about 4% of green H2 if renewable sources of energy are used (e.g., solar, wind), but it is limited by heavy efficiency penalties, and it may be suitable for a large market only if renewable electricity costs will be low enough. The steam reforming of glycerol [6,7,8], biogas [9], methanol [10], ethanol [11,12], etc., and gasification of biomass-derived feedstocks [13] are alternatives that are being investigated but need to overcome some technological barriers, many times related with the presence of contaminants. The photo-biological and photo-electrochemical methods are currently at a very early stage of development [2,5]. Figure 1 shows the main routes for H2 production.
H2 production through technologies that use green fuels, such as biomethane steam reforming or biomass-derived feedstocks gasification, have a relevant potential for industrial implementation [5,14]. In addition, comparatively with other renewable sources, it has a unique advantage, if the process is carried out with carbon capture and storage (CCS), carbon negative emissions could be achieved [15]. The technology readiness level (TRL), H2 cost, and CO2 emissions have a preponderant role in the large-scale implementation of these technologies. For example, the biomass-derived feedstocks gasification for H2 production (TRL 5-6) presents a very high decarbonization potential (~80% without CCS, compared with SRM without CCS) [2]. Moreover, negative CO2 emissions can be achieved if biomass-derived feedstocks gasification is replaced by gasification with CCS (TRL 3-5), i.e., −14.6 vs. 2.6 kg CO2/kg H2 [2].
Development of technologies with minimal environmental impact may decrease the world’s dependence on fossil fuels. Moreover, it can contribute to the decrease in political conflicts since the fossil fuel extraction is limited to very few countries [16]. The “in house” H2 production may increase both national energy security and contribute to economic development [17]. However, the cost of H2 production using renewable energy sources needs to be further reduced to ensure large-scale implementation [18]. Maggio et al. [19] believe that the first market of green H2 will be as feedstock for industrial applications, followed by power generation in stationary applications, and then the mobility sector, which presents major critical issues (cost, infrastructure availability, purity requirements, etc.).
Economic development, energy utilization, environment and climate are interconnected, but getting to “secure energy and environmentally friendly at lowest cost” is becoming increasingly difficult [20]. SMR is currently the most cost-effective process for H2 production, followed by coal gasification [21]. Large-scale H2 production by electrolysis of water with abundant electric power from renewable sources is expected in the long term; however, on a shorter term, due to a low level and insufficient global supply of renewable energy, H2 production from fossil fuels with CO2 capture and storage may prove to be an enabler for low CO2 emission H2 production [22] and ensure the transition needed for the future. Timely implementation of educational, financial, legislative, social and technological initiatives is necessary to make this happen.
Table 1 summarizes the main H2 production technologies, including their efficiencies, estimation of H2 costs, CO2 emissions and current technology maturity.
The commercial technologies based on the fuels reforming and gasification processes involve the production of syngas (H2, CO, CO2, and some CH4 and hydrocarbons). However, H2 purity has a high impact on end use applications, namely, in the case of fuel cells where H2 content in the feeding stream has to be above 99.97% [22]. The water–gas shift reaction is often used in industry to upgrade the syngas. Through the conversion of the CO into CO2, an additional quantity of H2 is produced [24], increasing the calorific value and composition of syngas. Separation techniques can then be applied for better hydrogen yields such as pressure swing adsorption (PSA), membranes, and distillation. Despite currently PSA being the most used technology with high levels of H2 purification, other advanced technologies such as membrane reactors, sorption-enhanced reforming or gasification and water–gas shift (WGS) reaction are in constant development.
The H2 production by WGS reactions is equilibrium-limited, but if CO2 is continuously removed, the reaction will be shifted in favor of increased H2 production (Equation (1)).
Depending on the temperature, during the reforming and gasification processes, the WGS reaction can contribute to an increase in the H2 production. As can be shown in Figure 2, H2 and CO2 formation takes place mainly at lower temperatures, which is not compatible with the operating conditions of the reforming and the gasification processes. Consequently, the syngas produced during the reforming or gasification (>600 °C) is upgraded in an independent WGS reactor (250–500 °C). The in situ CO2 capture during the reforming and gasification processes, or syngas upgrade in an independent reactor, can favorably influence the thermodynamic equilibrium of the above-mentioned reactions for a higher efficiency of H2 production and can lead to additional advantages such as increased energy efficiency, reduced reactor or catalyst volume. The concept of such advantageous combinations of process steps is known as process intensification. The combination of both processes, reaction and CO2 capture is defined as sorption-enhanced reforming (SER), sorption-enhanced gasification (SEG) and sorption-enhanced water–gas shift (SEWGS) reactions.
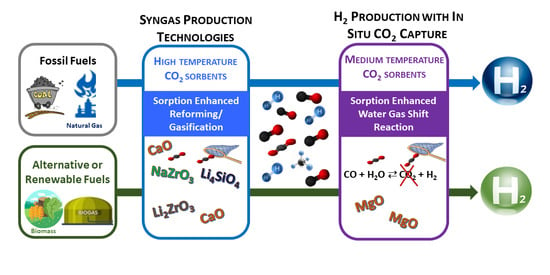
The selection of suitable CO2 sorbents, with high activity and stability for the sorption-enhanced processes, i.e., SER, SEG and SEWGS, will depend on the process reaction temperatures. Thus, the solid sorbents can be classified as high-temperature (>400 °C) CO2 sorbents (e.g., CaO, Li4SiO4, Li2ZrO3), which are mainly used for SER and SEG, and as medium-temperature (200–400 °C) CO2 sorbents (e.g., hydrotalcites, MgO) that are used for the SEWGS reaction. Some hydrotalcites can be used also at higher temperatures (~500 °C). Ideally, the sorbents should have the following properties: high mechanical stability, high adsorption capacity, fast reaction kinetics and low regeneration heat demand. In addition, if during the sorption-enhanced processes the CO2 sorbents regeneration generates pure CO2, two product streams can be obtained—H2 and CO2—which can also be valorized in downstream processes. The performance of solid CO2 capture sorbents has been largely studied and is described in the literature [26,27,28,29], but usually the research is focused only on the sorbent point of view. The goal of this review is to assess the sorbent influence on the reforming and gasification processes and during the WGS reaction, and also how the sorbent presence affects the H2 production yield. Only Ca-based sorbents, alkali-based sorbents and Mg-based sorbents will be considered since these are based on chemical reactions between the CO2 and the sorbent, while for example the CO2 capture by hydrotalcites is based on chemisorption processes. The Ca-based sorbents appear to be the most promising due to the fast carbonation kinetics and high theoretical CO2 carrying capacity, but the sorbents deactivation during the first’s cycles and the high energy consumption are drawbacks that need to be overcome. Currently, the alkali-based sorbents are not competitive due to the price, kinetic limitations and low CO2 carrying capacity. At medium temperature, the Mg-based sorbents present kinetic limitations, but the use of alkali molten salts to facilitate the CO2 diffusion on the sorbent show promising results. Detailed information about the sorbents and approaches to overcome some limitations can be found along the article. The catalyst-sorbent arrangement is important and may determine the success of sorption-enhanced reactions. Hybrid materials, i.e., with catalytic and CO2 capture activity, have also been gaining relevance, but the conclusions about their performance are often contradictory, and must be better understood.
Summarizing, aiming at increasing the H2 production yield, this review focuses on in situ CO2 capture materials, the high-temperature CO2 sorbents that are used mainly during the syngas production by reforming or gasification processes, and the medium-temperature sorbents are applied during the syngas upgrade through the WGS reaction.
2. High-Temperature CO2 Sorbents: Syngas Production
The syngas can be produced from a variety of feedstock sources, and it is a versatile intermediate gas obtained from reforming and gasification processes for the production of chemicals and fuels (Figure 3). The syngas composition is strongly dependent on the feedstock and the production technology (Table 2). The use of solid CO2 sorbents to improve the H2 content in the syngas obtained from the reforming and gasification technologies is described below.
2.1. Sorption-Enhanced Steam Reforming
Hydrocarbon reforming technology refers to the process of converting hydrocarbons into H2 using reforming techniques. Besides the hydrocarbon, the other reactants can be either steam (steam reforming) or oxygen (partial oxidation) or both (auto-thermal reaction) [16,34]. Among the hydrocarbon reforming, the SMR is particularly attractive due the level of maturity, high H2 production efficiency, and lower production costs (Table 1).
In the steam reforming process, hydrocarbon and steam are catalytically converted to H2 and CO2. The whole process involves the following major steps: generation of reforming or synthesis gas (syngas), water–gas shift reaction, and gas purification [21]. At the commercial level, the most usual raw material involved in the steam reforming process is methane from natural gas, but alternative raw materials containing gases through various combinations of light hydrocarbons including ethane, propane, butane, bio-methanol, bioethanol, biogas [9,35] are described in literature. The use of alternative raw materials such as biogas can be preceded by the H2S removal and/or biomethane enrichment to improve the H2 yield production and minimize catalysts poisoning [9]. The main chemical reactions that take place during hydrocarbon reforming are summarized in Table 3.
In a typical process, the steam and natural gas react at 850–900 °C, with pressures of 20–35 atm, and steam to carbon (S/C) ratios of 2.5–3.0, in the presence of a Ni-based catalyst to produce syngas. SMR is severely endothermic, and thus thermodynamically preferable under a high temperature and low pressure [34]. Lower temperatures increase coke deposition [22]. Despite the SMR being favorable at lower pressures, the methane activation by the catalyst is improved at high pressures, which justifies the high pressures used in industry.
The Ni-based reforming catalysts for H2 production face four major issues: loss of activity, poisoning, carbon deposition and sintering. The noble metals (Rh, Pt, Pd, Ru) have a better performance and coke resistance than Ni, but the high cost limits their utilization as catalysts for H2 production [36]. When the feedstock contains organic sulphur compounds, the reforming step should be preceded by a desulphurization step to avoid the catalyst poisoning. In the conventional systems, the syngas is fed into a WGS reactor, where the CO reacts with steam to produce additional H2, and then passes through a pressure swing adsorption system allowing one to obtain H2 with higher purity (near 100%) [21]. Besides in the SMR process there may be an additional CO2 production due to the furnace heating. If the SMR process includes CCS, a separate post-combustion CO2 capture unit is required for the flue gas. Despite the additional cost of SMR with CO2 capture, the overall production cost is still significantly lower that using renewable energy for producing hydrogen via water electrolysis.
The sorption-enhanced steam reforming (SESR) combines the reforming reaction system, i.e., reforming, WGS reaction and CO2 capture reactions, in a single reactor by the addition of a high-temperature solid sorbent. The in situ capture of CO2 overcomes the equilibrium limitations of the reforming and WGS reactions, through the shifting of the reaction equilibrium to the product side (Le Chatelier’s principle). The integration of reaction and product separation in a single step intensifies the process, increasing the H2 production yield and decreasing the size of downstream separation stages or even eliminating the need for them. At high temperatures (>600 °C), the most popular sorbents for in situ CO2 capture are calcium oxide, sodium and lithium silicate/zirconate-based materials. Some works with hydrotalcites are also described in literature.
The Ca-based sorbents are especially attractive for SESR due to the high exothermicity of the carbonation reaction. The heat generated during carbonation can be consumed in situ in the highly endothermic reforming reaction (Equation (2)), relieving the need for direct fuel combustion [26]. Moreover, the reaction enhancement enables lower operation temperatures (500–700 °C), reducing problems associated with high process energy requirements and poor energy integration within the plant environment, since the energy efficiency of the steam reforming process increases by approximately 20% [37]. Therefore, higher H2 production yields can be achieved while moving the optimum temperature of the reformer towards lower values compared to conventional SMR. Since the differences in the operating conditions of reforming and regeneration steps are considerable, the use of different reactors has been proposed to ensure continuous H2 production by periodically regenerating the sorbent [38]. This aspect will have an important impact on a potential scaling up of SESR technology to a commercial size. Currently, the SESR concept (TRL < 5) faces many challenges that can be overcome by the study of most suitable arrangement and operation of the reactors in various configurations, such as operating temperatures and pressures, reactors design, lifetime, cost of materials, etc. These studies are relevant to assess the economic, environmental, and technical feasibility of SERS. Figure 4 shows a simplified diagram of (a) SMR and (b) SESMR processes.
Emerging technologies to reduce the energy requirements of the SMR, and consequently the CO2 emissions, also includes the chemical looping hydrogen (CLH) process since the fuel is oxidized by oxygen provided by a solid oxygen material instead of mixing with air. The main chemical reactions of CLH are shown in Equations (5)–(7):
The major advantage of this process is that the required heat for converting CH4 to syngas is supplied without combusting part of the fuel. However, further studies need be conducted to improve the syngas quality, since it usually contains a substantial amount of CO2 because it is difficult to control the oxygen reduction carrier [37]. Nevertheless, Chisalita et al. [39] compared different configurations for H2 production by SMR with CO2 capture applying the CLH process and conclude that by using iron-based oxygen carrier, lower H2 production costs can be achieved comparatively with no capture conventional reforming (41.84 vs. 42.43 EUR/MWh).
2.2. Sorption-Enhanced Gasification
Gasification technology refers to the process of converting coal or biomass-derived feedstocks into gaseous fuel using a gasification medium such as air, O2 or steam [16,40]. It is a variation of pyrolysis that occur in inert atmosphere and, therefore, it is based upon partial oxidation of the feedstock material into a mixture of H2, CH4, CO, CO2 and higher hydrocarbons, i.e., syngas. Gasification is typically realized via the use of catalysts, which accelerate the rate of gasification and allow for the use of lower temperatures [18]. The catalyst efficiency is evaluated in terms of the H2 yield and is affected by the synthesis methods and the operation parameters. Ideally the catalysts should have a large specific surface area, high catalytic activity to effectively increase conversion rate and should be stable [14].
The critical factors that affect the syngas yield are the feedstock type, particle size, catalyst employed, operating temperatures (500–1400 °C), operating pressures (1 to 33 bar), and duration [16]. Some of the reactions that take place during gasification are:
In comparison with coal, the biomass gasification to H2 production is in an initial stage of implementation and facing problems related to feedstock distribution and technological aspects [5]. One of the problems is the production of tars, where the use of CaO acting as a CO2 sorbent also catalyzes the cracking reaction of tars enhancing the H2 purity and yield [17,41].
Sorption-enhanced Gasification (SEG) is being considered as a promising solid fuel conversion and CO2 capture technology that can produce H2-rich syngas mainly due to two reasons: (i) steam is used as the gasification agent [5,40] and (ii) removal of CO2 by the sorbent, both leading to an enhanced H2 production via WGS reaction [40]. The flow diagram of gasification and SEG are similar to the one shown in Figure 4a,b, respectively. However, depending on the sulphur content it can include a gas cleanup step before the shift conversion reactor [21].
In the Oxy-SEG, the air is replaced by O2 as oxidant agent, meaning that a flue gas rich in CO2 is obtained, making its storage or utilization easier. In comparison with the conventional gasification, the SEG and Oxy-SEG processes require lower gasifier temperature (600–750 °C vs. 800–850 °C) and produce high H2-rich syngas (>70% vs. <50%), higher H2/CO ratio (<10 vs. <2). The CO2 in the flue gas is lower than 10% in the conventional gasification process, and less than 30% or 90% in SEG and Oxy-SEG processes, respectively. This means that an optimized CCS/CCU can be attained for sorption-enhanced processes [42].
Mongkolsiri et al. [43] simulated the H2 production during biomass and coal co-gasification. The authors investigated the efficiency of Gasification-WGS and compared with the SEG-WGS and Gasification-SEWGS processes, between 500 and 1000 °C. Typically, a gasification process needs to be operated at high temperatures to achieve the required high H2 yield. The best performance was achieved for the Gasification-SEWGS process that presents a H2 concentration 45% higher than the conventional Gasification-WGS.
Fermoso et al. [44] stands out that the H2 yield increases with increasing temperature, which is justified by the enhancement of tar conversion, but the CO concentration can also increase probably due to the thermodynamic constraint of the WGS reaction at high temperatures. Therefore, selecting the appropriate operating temperature must be based on a compromise between the CO concentration and H2 yield. In the same work, it is shown that the H2 yield was not significantly influenced by the increase in S/C ratio from 2.3 to 9.4 during the sorption-enhanced steam gasification, performed with Pd/Co–Ni catalyst and dolomite sorbent, while it increased remarkably in steam gasification. These results underline the advantage of the SESG, as it allows the process to operate at a S/C ratio as low as 2.3 without significantly compromising the high H2 yield (76.1%) and purity (>98%). Consequently, SESG should significantly increase the energy efficiency of the process, reducing the operating costs due the use of a low S/C ratio.
2.3. Enhancement of the H2 Production with Ca-Based Sorbents
The use of Ca-based sorbents is considered a promising CO2 capture method for pre- and post-combustion conditions. This is due to the high theorical CO2 carrying capacity (0.78 g CO2/g CaO), the abundance and the low-cost of Ca-based sorbents that are available in nature such as limestone and dolomites, as well as the cost cut-off key feature of this CO2 capture process related to its carrying capacity and regenerative possibility. The cyclic carbonation–calcination is designed by calcium looping and is illustrated in Figure 5.
The calcination and carbonation reactions are both dependent on temperature, and its value is related to the CO2 vapor pressure as described by Equation (12) [45], and Figure 6 illustrates this dependence:
The calcination reaction (Equation (13)) at the calciner occurs at temperatures >900 °C (at 1 atm) and the CaCO3 is decomposed releasing the CO2 captured by the CaO, which would be recycled to the carbonator. This regeneration reaction is endothermic.
The atmosphere of calcination is a key aspect due the fact that for efficient compression, storage or utilization, the CO2 purity should be high; therefore, the calcination atmosphere ideally should be pure CO2. In the conventional configuration of CaL, an additional amount of fuel is combusted in an oxygen/carbon dioxide (O2/CO2) environment to maintain the desired operating temperature in the calciner and produce a high-purity CO2 stream. Both the temperature and the atmosphere parameters of calcination affect the sorbent morphology, i.e., specific surface area, total pore volume and CaO crystallite size properties suffer less modifications at lower values of temperature and CO2 partial pressure, which keeps the sorbent’s reactivity.
The carbonation reaction is exothermic (Equation (14)) and occurs at temperatures between 600 and 700 °C (at 1 atm), where CO2 is captured by CaO sorbent to form CaCO3, which in return is sent back to the calciner, and the cycle is continued. The high exothermicity of the carbonation reaction is an advantage in integrated processes since a high-grade heat can be utilized in the secondary power cycle for additional power generation.
The carbonation reaction occurs in two stages, a fast controlled stage (kinetic regime) that is dominant in the early cycles, and a slow diffusion limited stage (diffusional regime) that can become dominant after many cycles, mainly due to the loss of sorbent reactivity by sintering and pores’ blockage (Figure 7).
In the sorption-enhanced processes, the Ca-based sorbent loss of reactivity will have a negative impact on the H2 production. The key challenge of these materials is to minimize the sintering problems without significantly affecting the sorbent cost [46]. Approaches to increase the Ca-based sorbents lifetime include the use of inert additives for CO2 capture that can act as a solid support and/or to produce CaO-mixed oxides with a higher stability [47,48,49,50,51,52,53]. The change of sorbents morphology and microstructure through the use of different synthetic precursors and preparation methods [50,52,54,55,56,57,58,59] have also been studied. Selected inert additives should have or form oxides with a high Tammann temperature, contributing to the modification of the calcined sorbent skeleton/microstructure, hindering the aggregation or sintering of CaO crystallites, helping in the preservation of the nanocrystalline CaO structure [46]. Reactivation of sorbents with hydration, thermal and chemical pre-treatment is also reported. The use of solid Ca-based sorbents recovered from waste resources [60,61,62,63,64,65,66,67,68,69,70,71], as support or as doped materials [63,72,73,74,75,76,77,78] is recommended, due to its potential for circular economy.
Detailed reviews of Ca-based sorbents for CO2 capture, including sorbents carrying capacity, stability enhancement, and CaL applications were made by Sun et al. [26] and Salaudeen et al. [79].
The Ca-based sorbents regeneration is energy intensive due to the endothermicity of the CaCO3 calcination reaction, identified as a disadvantage for CaL, but it can be minimized through the addition of steam. The steam has a higher thermal conductivity than air or CO2, which allows for the use of lower calcination temperatures, minimizing sorbent sintering problems and fuel consumption. After the calcination, the steam can be easily separated from CO2 by condensation. More recently, the replace of conventional fuels by solar fuels had gained relevance, and some solar fluidized bed reactors’ prototypes for CaCO3 calcination are described in the literature [80,81,82].
Dang et al. [83,84] propose the integration of sorption-enhanced steam reforming of glycerol and methane reforming of carbonates to produce high-purity H2 without CO2 emission. The authors [84] introduce 1% of Pd in Ni-Ca-Al hybrid materials lowering the regeneration temperature of the hybrid material, from above 800 °C to 650 °C, since the Pd promotes the CH4 decomposition. Along the SESR of glycerol-methane reforming cycles, the catalyst showed a superior stability in terms of H2 production (98.5%) and CH4 conversion reaction (80%) because the Ni particle sintering was minimized. During the regeneration process, a syngas with a ratio of H2/CO < 3 was achieved for ten cyclic tests. A scheme of this new method is illustrated in Figure 8.
The SESR with Ca-based sorbents in a packed bed is usually divided into three characteristic stages [4,38,85]: pre-breakthrough stage, breakthrough stage and post-breakthrough stage (Figure 9). During the pre-breakthrough stage, the fresh sorbent provides timely in situ CO2 capture, which occurs simultaneously with steam reforming and WGS reactions. At this stage, almost pure H2 is collected at the reactor outlet. With the progress of CO2 capture, the capture rate of CO2 by the partially carbonated sorbents starts to decline, and the process shifts from a chemical reaction-controlled stage to a diffusion-controlled stage. Once Ca-based sorbents reach saturation, the CO2 capture becomes slow, and the breakthrough occurs. At this stage, the product H2 stream at the outlet starts to be mixed with some CO2. By further extending the CO2 sorption process, the sorbents will end up saturated and CO2 sorption rate would decline to zero. This is the post-breakthrough stage, which has no difference from traditional steam reforming. In an optimized SESR process, the pre-breakthrough stage should be extended as long as possible to allow for a suitable CO2 remotion and maintain a high level of H2 production. Once the breakthrough takes place, the reactor should be switched to regeneration mode.
Using favorable conditions, and a Ca-based sorbent mixed with steam reforming catalyst, Dou et al. [5] obtained a total concentration of CO2 in the product gas lower than 50 ppm. Regardless of the fuel and the operating conditions applied, the authors observe that the carbonation reaction can effectively shift the equilibrium of steam reforming and WGS reactions, overcoming the conventional processes limitations in terms of H2 production and feedstock conversion due to the thermodynamic boundaries of the reversible WGS reaction.
Mahishi et al. [85] performed steam gasification experiments (500–700 °C, 1 atm) with biomass (pine bark) in the presence of a Ca-based sorbent. The H2 yield, total gas yield and carbon conversion efficiency increased by 48.6%, 62.2% and 83.5%, respectively. This was attributed to the reforming of tars and hydrocarbons in the raw product gas due to the presence of CaO, meaning that the calcium oxide played a dual role of sorbent and catalyst. There are other authors [86,87,88] that use Ca-based compounds as catalyst–sorbent; however, some of them perform the experiments at temperatures above 800 °C. This means that, depending on the CO2 partial pressure, the equilibrium is being shifted for the CaCO3 decomposition zone (calcination reaction), so at those high temperatures the tars reforming by Ca-based materials are mainly responsible for the improved H2 production yields.
Wang et al. [89] produced H2 by catalytic conversion of biomass with and without in situ CO2 capture in a dual fixed bed reactor. The authors compare the H2 production using Ni catalyst supported in Al2O3 or in CaO and observe an increase in H2 production for Ni supported in CaO. It stands out that 500 °C was the highest temperature used during the experiments, meaning that probably higher H2 yields can be achieved if the optimal CaO carbonation temperature is used.
Shahbaz et al. [90] reported that the amount of Ca-based sorbents can also influence the H2 production. The CaO/biomass ratio from 0.5 to 1.42 has a positive impact on H2 production, syngas yield, carbon conversion efficiency, and gasification efficiency; however, for higher ratios a decrease in the H2 production was observed. The authors justify it with the carbon conversion efficiency and the gasification efficiency dropping to 29.2% and 38.2%, respectively, at a higher ratio (~2), since these parameters were measured based on moles of carbon-containing gases. However, there are disagreements in the literature about the optimal CaO/feedstock [91], which can be related with the feedstock properties and operating conditions. Detchusananard et al. [92] investigated the H2 production by the biomass SEG process and stated that a ratio of CaO/C > 3.2 should be maintained to ensure that the generated CO2 is completely captured.
For integrated gasification combined cycle (IGCC) power plants, CO2 capture can be implemented as either a post-combustion system for CO2 capture from flue gas, or a pre-combustion system for CO2 capture from syngas. However, the use of CaL as the pre-combustion CO2 capture configuration was found to result in an efficiency penalty 1–2% lower than conventional pre-combustion solvent scrubbing technologies [93]. Moreover, CaL has been shown to enhance the water–gas shift reaction, yielding a high-purity H2 stream, increasing the quality of syngas. To decrease the efficiency penalties of CaL associated to the endothermic sorbent regeneration, the solar-driven calcination in a fluidized bed reactor is an interesting alternative [81,94].
The cyclic CO2 uptake of Ca-based sorbents is a key factor that can limit the efficiency of SE processes. A reduction in the sorbent CO2 carrying capacity on cycling operation is regarded as the major challenge of pre-combustion CaL. As mentioned above, this happens because of sintering, pores blocking, but also due to sorbents attrition and sulphation. Specially in the case of gasification, the pores blocking can be more accentuated due to the tar species deposit on active sites of CaO surfaces. The modification of Ca-based sorbents to improve the stability and porosity along the time are the most promising approaches. Even so, to maintain acceptable sorbent conversion, the spent sorbent needs to be partially replaced by fresh sorbent. The ratio of sorbent/catalyst and the preparation methods can also affect the SESR and SEG processes’ efficiency levels. The in situ CO2 capture is influenced by the temperature, the steam and the pressure used during the SE processes. These conditions can be quite different from the post-combustion CaL conditions for CO2 capture.
2.3.1. Effect of Reforming/Gasification Temperature on Ca-Based Sorbents
Temperature is the most important parameter of the reforming/gasification process since it directly influences the conversion of feedstock and the CO2 capture. It stands out that the sorbents carbonation is strongly dependent on temperature and the CO2 partial pressure (Equation (12)).
During biochar gasification, Chimpae et al. [13] observed that the CO2 emission decreased with increasing temperature from 500 to 650 °C. The authors consider that it is justified by the enhanced CO2 sorption at 650 °C. Similar tendencies were observed by Muller et al. [95] during the wood SEG, i.e., the lowest CO2 and the highest H2 contents were obtained for temperatures between 630 and 750 °C. In line with this, during biomass gasification experiments with Ca(OH)2 as Ca-precursor, Guoxin et al. [96] observed that high temperatures go against CO2 capture, and identify the 650–700 °C range as the optimal operating temperature. Criado et al. [97] attributes the reduction in CaO carbonation to the slow diffusion of product molecules or ions at lower temperatures (<550 °C), resulting in a high density of small islands that cover the CaO surface more rapidly than at higher temperatures. Consequently, a smaller product layer thickness was formed, which reduced the sorbent’s carrying capacity.
In general, a consensus is found regarding operation in SEG, with temperatures lying between 600 and 750 °C. At a temperature above 750 °C, CO2 capture is not possible due to equilibrium constraints, and below 600 °C the CO2 capture rate is not feasible due to its slow sorption kinetics. Hence, for CaO to be able to adsorb the produced CO2, the temperature must be lower than that of the typical catalytic gasification range of 750–950 °C [91].
2.3.2. Effect of Reforming/Gasification Steam on Ca-Based Sorbents
It is well-established that the presence of steam enhances the extent of the carbonation reaction within a given time of Ca-based sorbents [29]. Donat et al. [98] noted that the steam affects the driving force (pCO2− pCO2,eq) needed for carbonation to be initiated. Morphological analyses of sorbents, after repeated carbonation–calcination multicycles, revealed that sorbents cycled in the presence of steam possessed a greater surface area and pore volume in the pore diameter range of 10–100 nm. The mesopores (2–50 nm) and small macropores (>50–100 nm) content have a positive effect on carbonation due to a lower diffusional resistance to CO2 along the sorbent when CaCO3 forms, which allows for higher CO2 uptake capacities over a given time.
Dong et al. [99] compared the in situ high-temperature steam (in carbonator/calciner) with the ex situ low-temperature steam (steam hydration in hydrator) and found that both arrangements are effective for CaO reactivation. For the first arrangement, the improved sorbent reactivity was mainly attributed to an increased pore volume, enhancing the extent of carbonation in the kinetic regime; in the latter, the formation of cracks accelerated the rate of diffusion. Considering the in situ high-temperature steam arrangement, Wang et al. [100] considered that the steam has a more relevant role during the diffusion-controlled stage than during the kinetic regime. The authors believe that the existence of OH− and H+ derived from H2O dissociation promotes CO3 2− formation. Due to its small size, the H+ diffuses easily through the CaCO3 product layer to the CaO/CaCO3 interface and reacts with O2− to form OH−, that will diffuse outward to the CaCO3/gas interface and react with CO2 to form CO32−, modifying the reactant structure and the reaction activity, leading to an improved carbonation [29,100].
Another possibility for the Ca-based sorbent carbonation enhancement could be related with the Ca(OH)2 formation, since this compound presents a higher reactivity than the CaCO3 [101]. Although Ca(OH)2 would not be stable at typical carbonation temperatures (>650 °C) and steam partial pressure, it may react only as an intermediate to enhance carbonation reaction, especially when lower reaction temperatures are used, through the following reversible reaction:
The intermediate formation and decomposition of Ca(OH)2 may also promote the structural rearrangement of the sorbent, leading to the development of a higher pore volume.
The steam presence during the calcination, apart from changing the sorbents morphology, also impacts the Ca-based sorbents’ regeneration because it decreases the CO2 partial pressure, and consequently lower temperatures are required during calcination. In addition, the higher thermal conductivity of steam comparatively with air or CO2 atmosphere also allows one to reduce the used calcination temperature. In fact, an equilibrium between the ratio steam/temperature needs to be found to avoid the sorbents’ sintering. There is not an agreement about the optimum fraction of steam in the calcination atmosphere. Donat [102] observed that the steam enhances the sorbent reactivity at concentrations as low as 1% with no significant improvement at higher concentrations. On the other hand, Champagne et al. [103] assessed the CaO conversion of two limestones in a TGA apparatus, without steam and with 5%, 15% and 40% of steam and obtained better conversions in the presence of 15% of steam. Later, using a pilot plant Champagne et al. [104] assessed the calcination of a natural limestone without steam (75% CO2, 20% O2 and 5% N2), with 15% of steam (balanced with 20% O2, 60% CO2 and 5% N2) and 65% of steam (balanced with 14% CO2, 20% of O2, 1% of N2). The authors have taken four samples from the carbonator during steady state operation and observed a higher and stable CaO conversion for samples tested with 65% of steam.
Donat et al. [98] schematized the relationship between sorbent morphology and conversion without steam, steam on carbonation, steam on calcination or both steps, which is presented in Table 4.
The steam also impacts on the fuel conversion during the reforming processes, which can affect the Ca-based sorbents’ performance. Usually, increasing S/C ratio, the H2 concentration is increased due to the enhancement of steam reforming and WGS reactions [42]. In addition, H2/CO ratio can be adjusted to some extent by variation of the S/C ratio, which can affect the sorbents’ performance. If during the reforming/gasification process a higher volume of CO2 is present in syngas, a faster regeneration or replacement of the Ca-based sorbent will be required.
For gasification systems, the steam is considered the most promising gasifying agent for H2 and syngas production. Shahbaz et al. [90] investigated the steam gasification of palm kernel shell in a fluidized bed reactor, in the presence of CaO sorbent and coal bottom ash as catalyst (692 °C), with a steam/biomass ratio between 0.5 and 1.5 (w/w), and obtained an increase from 35.8 to 79.8% of H2, respectively. This was explained by the degree of activeness of methane reforming, the water–gas shift and the char gasification reactions. However, depending on the catalyst/sorbent ratio, different types of feedstock and operating conditions can be found in the literature [91,105]. Tan et al. [105] obtained an enhanced H2 production using a Ni-Dolomite-La as catalyst/sorbent at 750 °C with a S/C molar ratio of 1. The authors observed by X-ray diffraction that for the steam reforming process at S/C ratio > 1, the Ni metal was oxidized. This decreased the production of H2, which can contribute to a higher deposition of coke in the catalyst/sorbent, reducing the CaO active sites’ availability and decreasing the CO2 sorption.
2.3.3. Effect of Reforming/Gasification High-Pressure on Ca-Sorbents
Thermodynamically, it would be preferable to conduct the reforming/gasification reaction under a high temperature and low/moderate pressures. However, high pressures are usually used because the streams coming out from the reactors need to be purified through successive processes, all of them requiring high operating pressures [106,107]. In addition, the high pressures speed up the reaction [34,107], and lead to a lower reactor’s volume, and subsequently lower costs [107].
The CaL process is usually performed at atmospheric pressure, as the high pressure adds further complexity to the process. As shown above, in Figure 6, the high pressures will be more favorable to carbonation, than to calcination. At the same temperature, the carbonation will be enhanced when the pressure increases; however, it has a negative effect in the calcination, since it will need higher temperatures, which contributes for the sorbent sintering. Butler et al. [107] summarized the advantages of CaL under high pressure: the carbonation reaction rates are improved, the operation of fluidized beds is more ‘smoothly’ with smaller bubbles and enhanced gas–solid contact, with enhanced heat and mass transfer. Hence, small reactor vessels can be used since greater molar gas flow rates occur at the same superficial gas velocity. On the other hand, the sorbent feeding becomes more challenging, and the entrainment of particles tends to be greater, more complex, expensive, and requires additional safety features.
Yu et al. [108] evaluated the high-pressure carbonation (700 °C) using three Ca-based sorbents, namely, CaCO3, Ca(OH)2, and PCC. The authors observed that the rate of the carbonation reaction initially increased with increasing total pressures up to 5.3 bar, beyond which the total pressure did not further enhance the reaction rate for all the Ca-based sorbents that were investigated. The carbonation reaction was found to be of 1st order at lower total pressures but changed to 0th order at higher total pressures. Furthermore, the carbonation reaction rate under pressurized conditions was found to decay more slowly than that under atmospheric conditions in multiple-cycle tests.
Shahid et al. [109] performed modelling studies on the sorption-enhanced steam methane reforming in an adiabatic packed bed reactor under low to medium pressure conditions (3–11 bar). The authors stated that the pressure increase had a negative effect on the CH4 conversion and the H2 yield, whereas the CO2 capture efficiency by the Ca-based sorbent increased, because the carbonation kinetics is favored by relatively high pressures. Using the Ca-based sorbent, the CH4 conversion was 82% and 72% at 3 and 10 bar, respectively. The H2 purity decreased from 85% to 84% as pressure increased from 3 to 4 bar. The decrease in H2 purity was justified by the decrease in the CH4 conversion from 82% to 77%, as pressure increased from 3 to 4 bar. Above 4 bar, H2 purity almost remained steady due to a slight increase in CO2 capture efficiency, which is in agreement with Yu et al. [108] observations. Ma et al. [38] reported that the negative effect of pressure rise on SMR was more pronounced than the positive effect of Ca-based sorbents in the carbonation. In addition, higher pressures sharply shortened pre-breakthrough duration and further lowered H2 purity.
2.4. Enhancement of the H2 Production with Alkali-Based Sorbents
In addition to Ca-based sorbents, the lithium silicate (Li4SiO4) [28,110,111], the lithium zirconate (Li2ZrO3) [110,111,112], and sodium zirconate (Na2ZrO3) [11,111] alkali sorbents are the most promising sorbents for CO2 capture at high temperatures. However, these sorbents are much less investigated in literature than the Ca-based sorbents, which can be partially justified by their kinetic limitations and lower theoretical CO2 uptake capacity. The alkali sorbents’ carbonation reactions, theoretical CO2 uptake capacity and operating carbonation temperature range are summarized in Table 5.
Among the alkali-based sorbents, the most studied is Li4SiO4. Figure 10 illustrates the carbonation–regeneration cycle.
The carbonation and regeneration reactions are both dependent on temperature, and it is noted that temperature is related to the CO2 vapor pressure, as described by Equation (19) [114] and illustrated in Figure 11:
However, the application of carbonation–regeneration reactions for sorption-enhanced processes is limited by the lithium high cost and by its slow kinetics during CO2 capture and regeneration, harming the H2 production and purity, especially for low CO2 partial pressures. In 2004, Abanades et al. [115] already argued that the performance of Li-based sorbents must be proven for up to ∼10,000 reaction cycles to be economically competitive with sorbents derived from those naturally occurring, such as limestone. The kinetic limitations are explained by the formation of a core-shell which delays considerably the carbonation/regeneration reaction. During the carbonation, the Li4SiO4 reacts with CO2 at the particle surface and forms an external shell that consists of Li2CO3 and Li2SiO3 (Figure 12). Then, the CO2 must diffuse through the external shell to react with Li4SiO4. At the beginning of the reaction, CO2 sorption reaction rate is controlled by the CO2 capture rate of Li4SiO4. However, once the carbonate-oxide external shell has been formed, the CO2 sorption reaction rate will depend on the diffusion processes of CO2, Li+ and O2− [113,116].
During the regeneration, it occurs the inverse process through the following reactions:
2Li2CO3 (s) → CO2 (g) + 2Li+(s) + O2− (s)
Li2SiO3 (s) + 2Li+ (s) + O2− (s) → Li4SiO4
First, the external layer of Li2CO3 releases the CO2, and the Li+ and O2− starts to diffuse through the intermediate Li2SiO3 product layer to form the Li4SiO4, but this diffusion process is slow, especially under a CO2-rich atmosphere. In order to improve the sorption performance of Li4SiO4, research interests had been focused on preparing the sorbent by different synthetic methods [117], or precursors [118], or doping the sorbent with molten salts [119], or doping with inert compounds [120]. However, the regeneration atmosphere is also a concern. Li et al. [121] investigated the effect of sorbent regeneration under 20%, 50% and 80% CO2 desorption atmospheres, and the results indicated that severe sintering occurs with the increase in CO2 atmosphere. For a large-scale implementation, these sorbents capture-regeneration properties need to be improved considerably [122].
The Li2ZrO3 sorbents exhibit CO2 sorption-regeneration properties similar to the Li4SiO4, since both sorption processes follow the double shell model. Moreover, these two sorbents have bad sorption capacity at a low temperature and low CO2 concentrations. For low values of CO2 partial pressure (~10%), the sorption capacity is almost zero, due to the constraint related with CO2 diffusion on the particle surface [123]. To minimize this problem, doping with molten salts or the formation of solid solutions by elemental substitution seems to be the most promising approach. The melting point of Li2CO3 is ca. 723 °C, but in the case of Li2CO3 (42.7%)/K2CO3 (57.3%) and Li2CO3 (52%)/Na2CO3 (48%) mixtures it is around 498 and 500 °C. This means that the formation of eutectics occurs in the carbonation range temperature. The formation of a molten carbonate phase will reduce the resistance of CO2 diffusion. The diffusivity of CO2 in the molten carbonate is ca. 10−5 cm2 s −1 at 500–600 °C [122], which is much faster than that in a solid carbonate. Thus, the CO2 sorption enhancement by Li2CO3–K2CO3 or Li2CO3-Na2CO3 doping can be attributed to the faster CO2 diffusion through the molten carbonate (Figure 13).
The replacement of Li by Na in the mixed zirconate oxides for CO2 capture at a high temperature appears to be an interesting option, since Na2ZrO3 sorbent is cheaper, and despite the formation of an external shell structure [124], it has shown faster absorption kinetics than the similar lithium-based oxides. However, the regeneration performance of Na-based oxides was worse than that for the Li-based oxides, and a high regeneration temperature is required [125]. The slow kinetics of regeneration was attributed to a slow diffusion of CO2 through a product layer of Na2CO3. Ochoa-Fernandez et al. [126] summarized the properties of the above-mentioned sorbents under SESMR process (Table 6).
For SEP with alkali-based sorbents, as happens with Ca-based sorbents (Figure 9), three stages for H2 and CO2 yields are observed: the pre-breakthrough stage, the breakthrough stage and the post-breakthrough stage. Among other factors, the duration of each stage will depend on the sorbent’s properties identified in Table 6. It is expected that the high pressure used in the reforming or gasification processes enhances the low kinetics of Li4SiO4, Li2Zr2O3, and KliZrO3 sorbents, but the studies performed at a high pressure are scarce. The research performed with higher CO2 partial pressures (>50%) also show improved CO2 uptakes [119,127], but these conditions are not adequate for reforming or gasification processes.
2.4.1. Effect of Reforming/Gasification Temperature on Alkali-Based Sorbents
In the case of alkali-based sorbents, the temperature effect on the sorption rate depends on both thermodynamic and kinetic factors. Kwon et al. [128] synthesizes sodium-based lithium silicate sorbents, a solid solution with Li3NaSiO4 and Li4SiO4, and observed an increase in CO2 uptake from 0.1 to 0.23 g CO2/g sorbent when the carbonation temperature increases from 450 to 550 °C, but the CO2 uptake decreased for higher temperatures. This is in agreement with the research performed by Essaki et al. [129], about the temperature effect on H2 concentration during the SESMR process, using Li4SiO4 sorbents. The authors analyzed the sorbent performance at 500, 550 and 600 °C, and observed that at 550 °C the H2 concentration was 93.6% and the CO2 was only 0.01%. This showed a good CO2 uptake from the sorbent at this temperature. At 600 °C, the CO2 uptake increased to 0.84%.
Sanna et al. [130] simulated the syngas composition of biomass steam gasification (50% H2O, 30% H2, 20% CO2) and assessed the CO2 uptake, at 380 and 500 °C, using a high-pressure thermogravimetric system (15 bar) and a lithium silicate material derived from a fly ash. The tests confirmed an improvement of CO2 uptake with the temperature increase, i.e., the sorbent capture was 0.05 and 0.12 g CO2/g sorbent, at 380 and 500 °C, respectively. The use of a fly ash as precursor can justify the low carrying capacity of this lithium silicate-based sorbent.
Xiong et al. [131] assessed the temperature effect in a TGA apparatus with Li2ZrO3 sorbent at 500, 575 and 650 °C, and confirmed that the kinetic stage has a duration of about 20–25 min, reaching CO2 uptakes of 0.11 g CO2/g sorbent for the first temperature and ~0.14 g CO2/g for 575 and 650 °C. Yi et al. [132] evaluated the temperature effect on CO2 uptake by Li2ZrO3 in a pilot-scale reactor and found higher CO2 uptakes than Xiong et al. [131]. Tests were carried out in a pure CO2 atmosphere at temperatures ranging from 450 to 600 °C. The sorbents showed a similar carrying capacity at 550 and 600 °C (~0.25 g CO2/g sorbent), but lower values at 500 and 450 °C, ~0.23 and 0.21 g CO2/g sorbent, respectively. The authors stated that with pure CO2, the reaction driving force, which is characterized by the difference between experimental and equilibrium CO2 pressure, does not change significantly. This is due to the fact that the experimental partial pressure of CO2 is much higher than the equilibrium CO2 partial pressure. This partial pressure driving force will be more dependent on temperature when lower experimental concentrations of CO2 are used. Lower temperatures should contribute to a higher CO2 uptake; however, this positive effect of lower temperature on the equilibrium CO2 partial pressure will be canceled by slow kinetics.
Xiao et al. [127] compared the uptake profiles of CO2 in K0.2 Li1.6 ZrO2.9 at a CO2 partial pressure of 0.25 bar and different temperatures (500, 550 and 575 °C) that are often operated, when the SE concept is applied in SMR-WGS reactions for H2 production. The CO2 uptake rate was slightly improved with increasing temperature, and the absorbed CO2 at the three different temperatures are almost the same (~0.23 g CO2/g sorbent), demonstrating its capability with different operating temperatures, when applied in SER.
Depending on experimental conditions and the type of the alkali-sorbent, enhanced CO2 uptakes can be achieved for temperatures between 500 and 650 °C. These sorbents will require a lower operating temperature during reforming and gasification processes than the Ca-based sorbents (600–750 °C), which can reduce the processes efficiency that are thermodynamically favored at high temperatures.
2.4.2. Effect of Reforming/Gasification Steam on Alkali-Based Sorbents
The steam effect on the CO2 capture is a relevant parameter for enhanced sorption processes for H2 production, since the most profitable processes include steam, i.e., steam reforming and steam gasification.
In the majority of the studies, the addition of a moderate amount of steam (∼10%) to the gas stream increased the rate of CO2 uptake at a high temperature, particularly during the diffusion-controlled stage, and improved the rate of regeneration of the sorbent [29]. Yi et al. [132] analyzed the steam effect on CO2 uptake by Li2ZrO3, using the following steam concentrations: 0%, 10%, 20% and 30%. The CO2 uptake increased with the amount of steam added, a higher improvement being observed between 10 and 20% of steam. After 120 min, the CO2 uptake was ca. 0.8, 0.13, 0.22 and 0.23 g CO2/g sorbent for 0, 10, 20 and 30% of steam, respectively. The authors believe that the steam media increase the Li+ mobility in the LiCO3 layer, improving the carbonation. The results suggest that Li2ZrO3 sorbent is more suitable for SMR or steam gasification than for post combustion capture, due to the high performance in the presence of high steam content.
Zhang et al. [133] compared the sorption capacity of K2CO3-doped Li4SiO4 sorbents, in a fixed-bed reactor, in the absence and presence of steam (dry atmosphere: 10% CO2, 90% N2; and moisture atmosphere: 10% CO2, 45% steam, 45% N2). The authors also observed an increase on CO2 uptake in the presence of steam, from 0.23 to 0.27 g CO2/g sorbent.
Ochoa-Fernández et al. [134] analyzed the effects of steam addition on the stability, CO2 capture and regeneration properties of Li2ZrO3, K-doped Li2ZrO3, Na2ZrO3, and Li4SiO4 sorbents. The authors confirmed that the increased steam addition initially improved the CO2 capture, except for the Na2ZrO3 sorbent. In this case, an improvement was observed when using 10% of steam, but the sorbent performance lowered with the increase in steam to 20 and 40%. Despite the improved CO2 uptake of sorbents in a steam atmosphere during the first carbonation, a large decay in the capacity was observed when compared to the performance of the sorbents under dry conditions, except for Na2ZrO3. After eight cycles, the CO2 uptake decreased from 0.23, 0.19 and 0.18 g CO2/g sorbent (0% steam) to 0.17, 0.02 and 0.07 g CO2/g sorbent (20% steam), for Li2ZrO3, K0.2Li2ZrO3.1 and Li2SiO4, respectively. In the case of Na2ZrO3, the performance of sorbents was almost constant after the 2nd cycle. The losses in capacity could be due to sintering under high steam pressure, to the vaporization of alkali metals, and/or phase segregation. The XRD analysis of the cycled sorbents showed that the alkali metal content was lower than before the cycling experiment for ZrO2-containing sorbents, whereas this was not observed for Li4SiO4 and K-doped Li4SiO4, reinforcing the occurrence possibility of the vaporization of alkali metals. Dunstan et al. [29] agree with the authors’ comments that the presence of steam can result in the hydrolysis of alkali carbonates forming molten alkali hydroxides at high vapor pressures (KOH > LiOH > NaOH), which are then evaporated gradually from the sorbent. The melting point of LiOH is ∼460 °C.
Further studies need to be conducted to understand the steam effect on the sorbent cyclability, to be able to evaluate the steam benefit for the alkali-based sorbents to increase hydrogen production yields.
2.5. High-Temperature Catalyst–Sorbent: Hybrid/Mixed Materials and Sequential Arrangement
The sorption-enhanced reforming and gasification processes usually use both catalyst and sorbents materials (exceptionally the same material can act simultaneously as a sorbent and a catalyst). Their performance is affected by the type of preparation, i.e., hybrid, or mixed materials. In the hybrid materials there is an enhanced contact between the catalyst and the sorbent; in this case, the sorbent can act as catalyst support, or the sorbent and catalyst can share a support material. The hybrid materials are also named dual or bi-functional materials. In the mixed sorbent/catalyst, both are physically dispersed with, or without, a sequential arrangement, and the contact between them is more superficial.
Many factors could affect the CO2 sorption performance and catalytic activity of hybrid materials, such as preparation methods, active metal components, the ratio of catalytic and sorption components, operating conditions of reforming reactions, etc. [4]. The hybrid material should present a good dispersion between the two active solid phases and the inert supporting material if it is present. Hybrid materials can face some limitations. For example, during the CaO carbonation, the sorbent is gradually transformed to CaCO3, and the porosity of the material will decrease due to particle densification. Thus, the availability of catalyst active sites will significantly reduce as a result of the outer shell of CaCO3 increasing volume, leading to a decrease in conversion into products and a complete deactivation [29,135,136]. Hence, the preparation methods should potentiate the active sites dispersion. Several synthesis methods were reported in the literature: co-precipitation [137], impregnation [111,135,136,138], mechanical mixing [139], wet–mixing techniques [135,136], and sol–gel methods [137]. The sol-gel methods, namely, those that the sorbent and catalyst are co-synthesized are usually promising, because the formation of homogeneous mixed oxides is favored [137], the dispersion of active sites is enhanced and the surface area is increased.
On the other hand, the proximity between sorbent-catalyst active sites can be an advantage because it improves the transfer efficiency of CO2 between the sorbent and the catalyst, since CO2 generated in the reforming reaction can be quickly captured by surrounding sorbents. Soltani et al. [140] considers that the hybrid materials have advantages, but more work is needed regarding life cycle, physical strength, attrition resistance, and sulphur tolerance. Another relevant point that was referred by the authors is the lack of information about their performance under high-pressure conditions as well as pilot-scale applications.
The optimization of sorbent/catalyst weight fraction is decisive to the performance of the SESR process. Giuliano et al. [138] synthesized CaO-Ca12Al14O33, Ni-Ca12Al14O33 and Ni-CaO-Ca12Al14O33 hybrid materials and tested them on a SESMR microreactor scale. The authors stated that with 30% and 54% of free CaO, the addition of 3% of Ni was not enough to maintain a stable CH4 conversion during the test, so neither a sorption-enhancing effect, nor a satisfactory reforming activity could be achieved. An increase up to 10% of Ni, on the same kind of sorbents, was necessary to improve the SESMR results. Later, Giuliano et al. [141] synthetized a CaO-Ca12Al14O33 hybrid material with 15% of free CaO on Ca12Al14O33, and 10% of Ni and tested the material in a SESMR microreactor scale during 3 h. The H2 production was constantly close to 90% in the pre-breakthrough period, while in the post-breakthrough stage it just had a nearly imperceptible decline from 77.0 to 76.5%, therefore very close to SMR thermodynamic equilibrium at the operating conditions. Correspondingly, CO2 in the output stream was constantly in the range 3.5–4.0% during the breakthrough, and between 10.5 and 12.0% in the post-breakthrough stage. The CH4 conversion was always higher than 95%. In fact, the chemical stability of hybrid materials is more challenging than for the case of mixed catalyst–sorbent systems, because of the coexistence of the active nickel, the calcium oxide and the support phases in the same particle.
The inert support plays an important role, stabilizing both catalyst and sorbent active functions, i.e., fuel steam reforming/gasification and CO2 capture. Therefore, simple and highly stable support formulations are essential [139]. The effect of sorbent support in the duration of the pre-breakthrough stage is also a relevant aspect.
Phromprasit et al. [142] synthetized Ni-CaO and Ni-Al-CaO materials by wet mixing; the outlet gas composition (H2, CH4, CO and CO2) indicated that the Ni-Al-CaO material exhibits a pre-breakthrough time longer than Ni-CaO. The stability of Ni-Al-CaO in the production of H2 over five cycles showed that CH4 conversion was >90% during the pre-breakthrough period for the five cycles. However, during the post-breakthrough period the CH4 conversion decreased slightly from 85% to 75% from the 1st to the 5th cycle, respectively. The authors justified this decrease by the reduction in active sites of Ni in the surface of the material. The XPS analysis showed a reduction in Ni concentration on the surface, from 1.93% to 1.02%, between the 1st and 5th cycle; this was caused by pore blockage due the CaCO3 formation.
The positive effect of inert support was also observed by Kim et al. [137]. The authors co-synthesized Ru/CaO and Ru/Ca3Al2O6–CaO materials by applying the sol–gel method and compared them with the following materials: impregnated Ru on limestone-derived CaO (Ru/lime) and a bifunctional Ni-based material (Ca–Ni-ex-HTlc). The Ru/Ca3Al2O6–CaO material showed an improved CO2 capture capacity (Figure 14a) and H2 production yield (Figure 14b), contributing to the enhancement of the SESMR performance, with an extended pre-breakthrough duration attained even after 10 cycles (Figure 14c). The Ru incorporation method was shown to be less relevant, since similar CO2 capture was observed for Ru/CaO and Ru/lime materials.
To overcome the CaO sintering problems during the SESMR process, Santos et al. [143] prepared Na2CO3-CaO sorbents and mixed them with 10% of Ni/Al2O3 catalyst. The hybrid material was tested for 10 cycles at 600 °C, showing 100% of CH4 conversion and a H2 molar fraction of 93.5%; however, the Na addition decreased the duration of the pre-breakthrough when compared with the non-doped material. This was explained by the increase in CaO crystallites in the presence of the alkali salt, which worsened the CO2 capture performance.
For SER processes, as illustrated in Figure 15, there are two typical configurations of catalyst/sorbent: alternated layers of catalyst and sorbent (Figure 15a,b), use of mixed (Figure 15c) or hybrid materials (Figure 15d) [144].
The packaging of alternated layers of catalyst and sorbent are considered a combination of several reforming reactors and CO2 adsorbers in series, where the catalyst/sorbent arrangement can affect the overall performance of H2 production. On the other hand, in the hybrid or pre-mixed materials, a well-mixed catalyst/sorbent can be produced, providing additional benefits related with the intensification of the heat and mass transfer processes compared to the conventional catalyst and sorbent arrangement [144]. Zhang et al. [133] reported that in the case of SMR, the uniform mixture of two materials has the ability of breaking chemical equilibrium since the in situ absorption packing is stronger than that obtained by the interval absorption packing of alternate layers, because the sorbent could not immediately remove CO2 in the last case.
Xie et al. [135] investigated the H2 production by SESMR in a fixed bed reactor using different patterns of catalyst (Ni0.5/Mg2.5Al) and sorbents (CaO–Ca9Al6O18). The comparison of small (0.16 mm) catalyst/sorbent pre-mixed particles and large (1.42 mm) pre-mixed particles showed a better conversion of CH4 with the small particles, which is explained by the strong mass transfer limitations of large particles. The authors also prepared large particles, with 1.42 mm, but integrating in the same particle the sorbent and catalyst (hybrid material) to evaluate if the integration of both into one particle could reduce the internal diffusion limitations. A similar CH4 conversion was observed to that in the pattern using the pre-mixed small particles during the pre-breakthrough period (~30 min). This is due to the CO2 that formed over the catalyst that could be directly captured by the sorbent in the same particle. However, after this period, the CH4 conversion decrease was more accentuated for the configuration with larger particles, even for hybrid material, due to the heavy internal diffusion limitations of reactants in the presence of a relatively large particle diameter.
Chimpae et al. [13] synthetized a NiO/Al2O3 catalyst, a CaO/Ca12Al14O35 sorbent and a NiO/CaO-Ca2Al4O35 hybrid material. These materials were used for biochar gasification. The H2/CO performance of gasification and SEG process show that the use of the hybrid material could provide the highest H2/CO, i.e., 0.18, 0.12–0.14 and 0.08, for hybrid material, opposite catalyst/sorbent configuration and without sorbent, respectively.
Wang et al. [145] analyzed the effect of internal diffusion resistances due the different configurations of sorbent–catalyst during the steam reforming of ethanol using Ni/Al2O3 and Li4SiO4 as catalyst and sorbent, respectively. In the first configuration, the catalyst and sorbent samples were uniformly premixed; in the second configuration, the catalyst particles were above the sorbent particles, and in the third configuration the sorbent and catalyst powders were placed at four-layer intervals with the catalyst on the upper layer. The last configuration shows that during the 10 cycles, the selectivity to H2 remains stable with hardly any fluctuations (>93%). Hence, there are a lot of factors that can affect the performance of sorbent/catalyst configurations, i.e., particle size, kind of sorbent or catalyst, sequential arrangement, sorbent and catalyst support.
For a better market penetration of sorbent–catalyst materials in reforming/gasification processes for H2 production, the performance of both, considering different configurations, needs to be further studied on a pilot scale. Foreseeing the sorbent–catalyst upscale potential, the cost-efficiency of the materials can be a crucial aspect. Thus, the use of wastes as precursors can be an alternative to reduce the costs. In the case of Ca-based sorbents, the CO2 carrying capacity of sludges from pulp and paper industry and some species of biomass ashes can be evaluated. The use of recovered lithium from ores, brine, sea water or recycled batteries [146] to synthesize Li4SiO4 or Li2ZrO3 sorbents precursors is an alternative that can be explored. The use of nickel recovered [147] from industrial wastes or spent batteries to produce catalysts is also suggested.
Table 7 compares the H2 production obtained using a conventional and sorption-enhanced processes. Different feedstocks, sorbent/catalyst composition and patterns, technologies and experimental conditions are considered. A sorption enhancement indicator is proposed to compare the H2 production enhancement due the sorbents’ capture that measures the increase in H2 production, , where SEP and CP are related to sorption-enhanced processes and conventional processes, respectively. An improvement in the H2 production is observed for both technologies, steam reforming and gasification when the sorption-enhanced processes replace the conventional processes, i.e., the H2 production has an increase between 6 and 55% and 31 and 148%, respectively. SESMR processes often achieve production values of H2 > 90%, which makes this technology very promising.
The SEP processes present several benefits that are summarized in Table 8, but some challenges need to be overcome for their application on a large scale.
3. Medium-Temperature CO2 Sorbents: Syngas Upgrade for Better H2 Yields
3.1. Sorption-Enhanced Water–Gas Shift Reaction
The water–gas shift reaction (Equation (1)) using syngas feedstock derived from coal gasification or steam methane reforming is a very common means of generating pure H2. The syngas composition depends on several factors, such as the selected reforming or gasification process, the fuel composition, the S/C ratio, temperature and pressure conditions of the shift reaction. Usually, syngas that originated from natural gas has a higher H2 and a lower CO2 and CO content than coal-derived syngas (Table 2). Besides, it has very low levels of sulphur compounds because most of it is removed before the reformer to prevent catalyst poisoning, while coal-derived syngas typically contains more sulphur. As mentioned in the above sections, the WGS reaction can contribute to the syngas upgrade by converting CO into CO2, which can be easily separated, and contributes to an additional production of H2.
At low temperatures, the WGS reaction is thermodynamically favored, since its equilibrium constant decreases as temperatures increases [158], but not kinetically, as it is an exothermic reaction (Equation (1)). Since the WGS reaction proceeds without change in the number of moles, pressure does not affect equilibrium, but up to the equilibrium moment total pressure can positively affect CO conversion since it increases the reaction rate [159,160]. To overcome thermodynamic and kinetic aspects, WGS reaction is carried out industrially in two steps: the first, high-temperature shift (HTS; 350–500 °C), using Fe-Cr catalysts, and the second, low-temperature shift (LTS; 200–250 °C), using Cu-Zn-Al2O3, that allows for achieving CO concentrations near 3% and 0.1%, respectively [161]. However, these two steps increase the complexity and the energy requirements of the process. Besides, if O2 is present it acts as a poison for the HTS catalysts, due the oxidation of Fe3O4 into the inactive Fe2O3 [37]. In addition, the LTS catalysts require high-volume reactors and can lose activity easily, due to being susceptible to poisoning by S, and Cu sintering. If H2S is present, a cobalt-molybdenum catalyst should be used [162]. On account of the limitations of commercialized shift catalysts, some improvements have been developed, such as replacing part of the metals with modified materials or doping with some alkalis [158]. Pal et al. [24] considered three more catalysts’ categories, ceria and noble metal-based catalysts, carbon-based catalysts and nanostructured catalysts [163]. The WGS reaction requires a step for the CO2 removal, usually the pressure swing adsorption technique is used.
In recent years, the sorption-enhanced reaction (SER) has been studied, aiming to improve the performance of the WGS reaction and leading to SEWGS reaction. The SEWGS reaction consists of a WGS reaction with in situ CO2 capture occurring simultaneously in a single reactor. According to Equation (1), the implementation of in situ CO2 removal shifts the WGS reaction to its right side by capturing the CO2 and, thus, increasing the H2 yield. That is to say that in situ CO2 capture overcomes the limitations related to the equilibrium, resulting in both higher CO conversion and enhanced H2 production. The result is the production of a very-high-purity H2 without the elevated costs associated to a separation process and a much more compact and simple process, associated with a higher energy system efficiency and lower capital costs. Hence, SEWGS is considered a promising pre-combustion CO2 capture technology [144,164,165,166]. Figure 16 schematizes the WGS and SEWGS systems’ equilibrium.
The success of the SEWGS is highly dependent on the sorbent selected for the in situ CO2 capture from the reaction medium. The adequate materials for this technology are medium-temperature solid sorbents. The hydrotalcite and modified hydrotalcite-based sorbents are largely studied [12,168,169,170], and exhibit low CO2 sorption capacity, i.e., less than 0.1 g CO2/g sorbent at medium temperatures, which in SEWGS will cause CO2 saturation and reduction in H2 production. Mg-based sorbents have also been applied in the enhanced CO2 removal from WGS reaction, having a high theoretical carrying capacity, which makes it very attractive. Moreover, the integration of Mg-based sorbents allows one to remove the CO2 at medium temperatures, that is, ranging from carbonation to calcination temperatures of 300 to 450 °C, respectively, without the need of low-temperature WGS reaction. The performance of SEWGS reaction in this range of temperatures can be greatly enhanced in the presence of suitable catalysts, i.e., with enhanced stability, applicability, and activity at medium temperatures [24]. The catalytic performance depends on the synthesis technique, the nature of the active site/phase, the type of support and the reaction environment [24,171,172,173]. At medium temperature, for single-stage WGS reaction, platinum-based catalysts and supports with oxygen storage capacity, such as CeO2, are particularly favorable, because they show very high activity [159,174]. In practical applications, Mg-based sorbents are promising; however, more studies need to be conducted to approach the theoretical carrying capacity. This type of sorbents show an abrupt decrease in the CO2 carrying capacity under long-term cyclical operation, sintering, attrition, and potential competing sulphation reactions [27,165,166,167]. In fact, there is no record of pilot-scale projects in this area [167], but promising results have been obtained using Mg-based sorbents modified with alkali molten salts [165,166].
3.2. Enhancement of the H2 Production with Mg-Based Sorbents
In recent years, there has been a significant growing interest among scientific researchers in using Mg-based sorbents for CO2 capture [175]. There are various benefits associated to Mg-based compounds. Mg-based sorbent is nontoxic, noncorrosive and is largely available on nature, where it is abundant at a relatively low cost. It also offers a wide temperature range to work with, from room-to-medium temperatures. Its regeneration can occur below 500 °C, which is a moderately low temperature, when compared to the one used with high-temperature CO2 basic metal oxide sorbents, such as Ca-based or alkali-based sorbents. Economic benefits unfold from using lower temperatures, since energy consumption is reduced, which in turn impacts positively in the system efficiency and the use of energy resources. In addition, Mg-based sorbents have a moderate basicity, which leads to a high theoretical CO2 capture capacity of 1.09 g CO2/g MgO. One mole of MgO can absorb one mole of CO2, according to the reversible reaction described in Equation (22) [175].
In practice, most of Mg-based sorbents do not exhibit the expected theoretical capture capacity. In fact, these materials are characterized with low CO2 capture capacity due to having slow kinetic reactivity [27,175]. As an example, commercial MgO powder presents a CO2 capture capacity of only 20 mg CO2/g MgO at 200 °C [176]. There are two main factors that are believed to explain both the poor capture capacity and the slow kinetics. Regarding the first one, this is related to the fact that MgO has a low surface area and, hence, does not expose its basic sites sufficiently well for CO2 sorption. Taking the same example of the commercially available MgO as above, its surface area is between 8 and 35 m2/g [27]. Moreover, MgO has a volume expansion of 2.49 times [27] caused by the formation of MgCO3. This product layer ends up covering with dense layers the adjacent basic active sites of the MgO sorbent, inhibiting the CO2 sorption to proceed. This evidence supports the fact that the poor sorption capacity is a surface phenomenon [175]. The other reason lies in MgO’s intrinsically high lattice enthalpy. Low porosity is also often related to low kinetics, since MgO’s strait pores obstruct the CO2 diffusion through them and, thus, delay the adsorption equilibrium [175]. In addition, MgO has a poor thermal and mechanical stability.
The circulation of the Mg-based sorbent between both the carbonation and the regeneration reactors is illustrated in Figure 17.
In order to understand the thermodynamic limitations that lead to constraints of the operational conditions associated to the equilibrium described in Equation (22), the CO2 partial pressure, PCO2, in function of the dissociation or equilibrium temperature, Teq, is plotted in Figure 18; this is calculated using Equation (23) [177].
The thermodynamic equilibrium of MgCO3 decomposes itself in MgO and CO2, at 1 bar and above 300 °C; the CO2 pressure represents a limitation in what concerns increasing the working temperature [178]. Only if working at higher pressures would higher operating temperatures be possible to consider, but both kinetic and uptake could still be a concern and a limitation for practical applications [176,178]. The possibility of working at relatively low temperatures makes MgO compounds attractive to explore, especially for SEWGS reaction, but it is mandatory to overcome uncompetitive capacities and low sorption kinetics rates first. In this sense, researchers started to study different paths to enhance their performance, based on the sorbents’ dependence on intrinsic and extrinsic factors.
Most promising approaches aiming to improve the CO2 capture performance of Mg-based sorbents by upgrading their internal properties consist of the following: synthesizing mesoporous MgO, producing MgO from effective magnesium precursors, dispersing MgO on inert supports and doping MgO with alkali molten salts (AMS). The doping with alkali metal salts is the most widely recognized promising approach [175].
The aim of the most recent experimental works is to improve the CO2 uptake capacity of these materials up to 0.7–0.8 g CO2/g sorbent [179]. The alkali carbonates and the alkali nitrates/nitrites are the most commonly used [27]. In general, three categories of alkali doping are considered: alkali carbonate doping, alkali nitrate/nitrite doping and binary or ternary alkali doping.
Regarding alkali carbonate doping, the CO2 mechanism sorption is believed to happen in two steps. The first step consists in the quick generation of basic sites on the MgO surface, due to the established interactions between the sorbent and the alkali metal carbonate molecules. The nature of the AMS highly impacts the kinetics and the sorption capacity of the doped MgO sorbent at this stage because the basicity level of the produced active sites is influenced by the size of the ion salt. The second step is the slow formation of the double carbonate phase between the Mg and the AMS [27]. Concerning the alkali nitrate/nitrite, it was shown by Zhang et al. [180] that a MgO sorbent doped with NaNO3 exhibiting good CO2 sorption kinetics and a MgO conversion of 75% against of only 2% for an undoped MgO, both at 330 °C and ambient pressure. It was stated that molten NaNO3 provides an alternative reaction pathway to traditional gas–solid reactions, by acting as a phase transfer catalyst between bulk MgO and CO2 molecules which, in turn, facilitates the sorption reaction. It was described as the promoting effect of the molten nitrate. In addition, molten alkali metal nitrates are shown to prevent the formation of a rigid, CO2-impermeable, and monodentate carbonate layer on the surface of MgO as it occurs with bare MgO, but to promote the rapid generation of carbonate ions to allow a high rate of CO2 uptake. The binary doping with alkali nitrate/nitrite is also an interesting matter of study. Zhao et al. [181] compared the CO2 sorption capacities of the single NaNO3 and of the binary NaNO3/NaNO2 doped MgO sorbents. The latter showed higher CO2 sorption capacity than the former. This new evidence found explanation on the reduction in the melting temperature of the eutectic mixture. While single NaNO3 and NaNO2 present a theoretical melting point of 308 and 271 °C, respectively, the eutectic mixture of NaNO3/NaNO2 exhibits a melting temperature of 185 °C. Thus, the eutectic mixture facilitates the sorption process by providing a molten phase that works like a liquid channel. Ternary doping with NaNO3, lithium nitrate (LiNO3) and potassium nitrate (KNO3) registered an even more accentuated reduction in the eutectic mixture’s melting point and an enhanced CO2 sorption performance. In the case of the ternary doping with LiNO3, NaNO3 and Na2CO3, the former two form the molten phase in which Na2CO3 dissolves along with the bulk MgO to react with the CO2 molecules [27,181]. It is well accepted that the melting temperature of the eutectic mixture impacts greatly on the CO2 sorption performance.
The enhancement of Mg-based sorbents carrying capacity boosts its use for SEWGS processes, but the studies available in literature, considering the simultaneous WGS reaction and CO2 capture are scarce, but promising.
To the best of our knowledge, the first experimental work conducted with Mg-based sorbent in a SEWGS reaction was performed by Abbasi et al. [182]. The authors tested a partially calcined dolomite impregnated with K2CO3 as sorbent/catalyst, at 20 atm, in a simulated syngas atmosphere. The sorbent was shown to be capable of achieving 95% of CO2 capture and 40% of conversion in the WGS reaction, but both activities decreased with increasing temperature. The results indicated that the pre-breakthrough WGS conversion diminishes as the sorbent is carbonated and CO2 concentration approaches the inlet concentration, leading to the conclusion that the catalytic activity of MgO is significantly greater than that of MgCO3. During the SEWGS at 400 °C, the H2 (dry basis) change from ~60 to ~45%, and CO2 from ~9 to ~25%, in the pre- and post-breakthrough phases, respectively.
Hu et al. [165] synthetized AMS-promoted MgO-CaCO3 sorbents and obtained a high CO2 carrying capacity and stability after 30 cycles, i.e., 0.55 g CO2/g sorbent (carbonation at 350 °C, 30 min, 1 atm, 50% CO2; and regeneration at 420 °C, 10 min, 1 atm, N2). The enhancement of sorbent performance resulted in a high H2 purity during the SEWGS process. For the optimized conditions, i.e., 12 atm, 300 °C, an initial ratio H2O/CO molar ratio of 1.5 and a three catalyst/sorbent layered configuration, the H2 purity was 99.4% for the 1st cycle and 98.2% after 10 cycles. In a more recent work, Hu et al. [183] describes the preparation of K2CO3-promoted Cu/MgO-Al2O3 by sol-gel method to be used in a SEWGS reaction. Very much promising results were obtained for a sorbent with a K/(Mg + Al) ratio of 0.2 and a Mg/Al ratio of 9. A H2 yield of 99.9% was registered after 10 SEWGS/regeneration cycles at 300 and 380 °C for SEWGS and for regeneration, respectively.
In another work, Lee et al. [166] reported a Na-Mg double salt-based sorbent that was tested under SEWGS conditions using a commercial catalyst. A divided section packing concept of catalyst/sorbent was prepared and a high pure H2 was obtained (CO < 10 ppm). The carrying capacity of this Na-Mg double salt-based sorbent was ca. 0.15 g CO2/g sorbent, so the reactor column was divided into more sections (~10) and packed with increasing amounts of sorbent.
3.2.1. Effect of Temperature on Mg-Based Sorbents
As shown in Figure 18, the MgO carbonation is strongly dependent on temperature, but the sorbent synthesis and properties, such as the use and type of promotors, also had a relevant role on the CO2 uptake. Wang et al. [176] analyzed the effect of temperature on CO2 sorption by NaNO2 and NaNO3-promoted MgO. It was observed that at low temperatures (240–260 °C), the 0.2NaNO3/MgO sorbent exhibited relatively low CO2 uptake. On the other hand, the 0.2NaNO2/MgO demonstrated faster weight increases, which indicates that the formed MgCO3 product layer of 0.2NaNO2/MgO is thicker than that of 0.2NaNO3/MgO, which increases the CO2 diffusion resistance. Further increasing the temperature to 280–300 °C, the CO2 sorption of two sorbents was significantly enhanced since the diffusion process was activated, and values were attained near 0.55 g CO2/g sorbent after 60 and 120 min for 0.2NaNO2/MgO and 0.2NaNO3/MgO, respectively. With the temperature increasing to 320–340 °C, the sorption rates decreased during the initial period, whereas the final uptakes were slightly improved. It was justified by the increased CO2 equilibrium concentration in the molten salts and the enhanced diffusivity of CO2 in the product layer with increasing temperature. Hiremath et al. [184] synthetized KNO3-LiNO3/MgO-TiO2 sorbents and observed that the CO2 uptake initially increases with increasing temperature from 250 to 300 °C, and started to decrease for higher temperatures (325, 350 and 375 °C), which is in line with previous results. The kinetics of CO2 uptake showed an interesting behavior at a lower temperature (250 °C): the CO2 uptake was fast at the beginning (<10 min), but at 300 °C the initial 10 min showed a slight increase in CO2 uptake followed by a fast transition leading to a higher CO2 uptake, although the initial sorption kinetics was slower. Wang et al. [185] found similar behavior with the sorption temperature, i.e., during the initial stage of the CO2 absorption process, the lower the temperature (275 vs. 375 °C) the higher the rate, which was justified by the higher CO2 concentration in the molten salt at a lower temperature. With the progress of CO2 absorption, the disadvantage of slow kinetics at low temperatures is more prominent, and a low CO2 uptake after 120 min of absorption was observed. Pozzo et al. [179] analyzed the cyclic performance of MgO promoted by 10% of (Li,Na,K)NO3 for different carbonation temperatures, 250, 275 and 300 °C, with a partial pressure of CO2 of 0.2 atm. At 275 °C, the CO2 uptake was higher, which was explained by the higher thermodynamic driving force at lower carbonation temperatures. The authors state that the eutectic mixtures become particularly important, as the low melting point broadens the operating window of the material.
Then, the temperature affects the kinetics that is a relevant aspect of the in situ CO2 uptake. The Mg-based sorbent carbonation should be quick enough to produce high-purity H2 during the SEWGS process.
3.2.2. Effect of Steam on Mg-Based Sorbents
The SEWGS process requires the presence of high quantities of steam for the WGS reaction, which justifies the understanding of the steam effect on the Mg-based sorbents performance. Zarghami et al. [186] investigated the effect of the presence of H2O on the reactivity of Mg-based sorbents. The experimental results demonstrated that the existence of steam in the sorption step had a positive influence in the rate of the carbonation reaction. The authors carried out several tests using reactant gas mixtures containing 50% CO2 and increasing concentrations of steam (10, 20 and 30%), in a pressurized system (20 bar) at 430 °C. A positive relationship was observed between the steam increase and the CO2 uptake, attaining values near 100% of CO2 uptake with 30% of steam, after 15 min, as shown in Figure 19.
Water is believed to work as a co-sorbent that boosts CO2 chemical reactivity, by creating a new carbonation pathway consisting of two mechanisms [175]. The primary mechanism forms an alternative transient compound, Mg(OH)2, with a larger molar volume than that of MgO (Equation (24)). The secondary mechanism acts at the pore structural level, by expanding the inner pore volume and, thus, diminishing the resistance through diffusion in its inside (Equation (25)) [27,175]. The overall result is the higher reactivity of the Mg-based sorbent toward CO2, that is, higher CO2 uptake capacity.
The kinetics of the reaction between CO2 and Mg(OH)2 described in Equation (25) is faster than that of the reaction between CO2 and MgO described in Equation (22). The capture of CO2 with Mg(OH)2 is an exothermic reaction with a ∆H value much lower than that of the reaction of MgO with CO2, i.e., −19.7 vs. −100.9 kJ/mol. Moreover, the capture of CO2 with Mg(OH)2 at a high temperature is faster than with MgO. Researchers deduce that the presence of water provokes the rearrangement of surface oxide to hydroxide over MgO molecules, producing Mg(OH)2. This transient species have weaker lattice bonds when comparing to MgO, which smooths the transfer ability of OH− more than O2−.
At atmospheric pressure, the Mg(OH)2 registered an absorption capacity of 0.148 g CO2/g sorbent (1 bar), but its operation is limited to the temperature window of 200–315 °C and requires the rehydroxylation of MgO in the sorbent regeneration [176,178]. In addition, the existence of water decreases the operating temperature. Thus, the regeneration of MgCO3 into Mg(OH)2 can be achieved at lower temperatures [27,181,182].
Yang et al. [27] also found that the presence of H2O during the sorption step improved the kinetics of sorption rates. In addition, it is also reported that the introduction of H2O at the desorption step could have benefits in the improvement of both regeneration rate and efficiency of Mg-based sorbents. These results support the idea that the steam will be beneficial to the performance of SEWGS process when Mg-based sorbents are used for CO2 capture, especially at high pressures.
3.2.3. Effect of Pressure on Mg-Based Sorbents
Currently, SEWGS processes take place at high pressures [27]. Thus, a CO2 separation unit of a SEWGS process demands Mg-based sorbents to keep its cyclic CO2 uptake capacity stable at high pressures. Hwang et al. [164] investigated the effect of the operation pressure in the CO2 uptake capacity of a Mg-based sorbent impregnated with alkali metal nitrates under multiple cycles. The obtained experimental results showed a general upward profile of the CO2 uptake capacity with increasing operation pressure: the CO2 uptake capacity increased dramatically from 1 to 20 atm, while a more discreet increase was registered from 20 to 30 atm. This fact was attributed to gaseous diffusion being mainly controlled by Knudsen diffusion at higher pressures. A Mg-based sorbent impregnated with 5% of NaNO3 plus 5% of KNO3 was able to maintain its CO2 uptake capacity at 0.40 g CO2/g sorbent after five cycles at 300 °C and 20 atm. It was concluded it was an adequate sorbent to be used in a SEWGS process at high pressures.
Hu et al. [165] analyzed the pressure effect on the outlet gas composition at the pre-breakthrough stage of a SEWGS process, using an AMS-Mg95Ca5 sorbent and increasing the total pressure from ambient pressure to 12 atm. The authors observed that the outlet concentration of CO2 during the pre-breakthrough period decreases, whereas those of H2 and CO increase, which is reasonable because the driving force for CO2 sorption increases with total pressure (Figure 18). For 1, 4, 8 and 12 atm, the measured CO2 concentrations during pre-breakthrough were 13.6%, 3.3%, 1.9% and 0.8%, respectively, while the equilibrium values were 5.2%, 1.3%, 0.6% and 0.4%, respectively. Therefore, the higher the pressure, the nearer the concentration approaches the equilibrium value. At the post-breakthrough stage, where the sorbent is completely saturated, no CO2 sorption occurs and only WGS takes place. At this stage, the outlet concentrations of H2, CO2 and CO at different pressures tend to the equilibrium values (46.3%, 46.3% and 7.4%, respectively)
Ryu et al. [187] examined the CO2 absorption properties of MgO-based sorbents loaded with K2CO3 according to the pressure (1, 10 and 20 atm). The MgO-based sorbent loaded with K2CO3 showed improved CO2 capture capacity at higher pressures, which was attributed also to the reaction of MgO and K2CO3 in the presence of water vapor at 20 atm, namely, due the formation of structures, such as MgCO3∙H2O and K2Mg(CO3)2. Hence, a positive effect of the high pressure on the CO2 uptake during the SEWGS process is expected.
In agreement with the MgCO3 thermodynamic equilibrium relative to CO2 partial pressure, it was reported [27] that working at a higher desorption pressure results in a higher desorption temperature needed for the regeneration of the sorbent. Hwang et al. [164] investigated the regeneration capacity of an AMS-promoted Mg-based sorbent at a high pressure and 100% CO2 condition. The authors observed that the CO2 gas was desorbed at temperatures above 575 °C, with a peak at about 620 °C for CO2 desorption at 20 atm and 100% CO2. Based on this result, it was concluded that the optimum regeneration temperature was greatly shifted from 430 to 620 °C when regeneration conditions of 100% CO2 were used, compared to when N2 gas was used.
3.3. Medium-Temperature Catalyst–Sorbent: Hybrid/Mixed Materials and Sequential Arrangement
The application of WGS catalyst—Mg-based sorbents—during the SEWGS reaction is recent, and few experimental studies contemplate its use. Lee et al. [166] studied the influence of the catalyst packing method in the CO2 removal of a WGS reaction, using a Na-Mg double salt as sorbent and a commercial catalyst (Cu/ZnO/Al2O3). In a first attempt, it was successfully synthesized a one-body hybrid solid by ball-milling, consisting of both catalyst and sorbent. However, it exhibited low sorption capacity when compared to that of single materials. The XRD analysis shows that the characteristic peaks for NaNO3 in the one-body hybrid solid disappeared after the SEWGS reaction. Since the NaNO3 crystalline structure was not recovered after cooling, the formation of the molten phase was inhibited, resulting in poor CO2 sorption. It may be possible that the reduced Cu reacted with oxygen, converting NaNO3 to NaNO2. Subsequently, a multi-section concept was adopted for the reactor that minimized the contact between the catalyst and the sorbent. This attempt generated high-purity H2 by SEWGS. Moreover, higher production of high-purity H2 (>98%) was registered when using a higher ratio of sorbent-to-catalyst, as higher concentrations of CO2 were being captured. It was also observed that the SEWGS performance improved with the increasing number of the reactor sections. The effluent gas composition from the SEWGS reaction in a ten-section reactor and a total catalyst-to-sorbent ratio of 1 (1 g of catalyst or sorbent) were alternately loaded in each section. It showed that the pre-breakthrough of CO2 and CO was ∼25.5 min. Further investigations using a reactor packing method with different catalyst-to-sorbent ratios were proposed.
The SEWGS experiment performed in a fixed bed reactor using AMS-promoted MgO-CaCO3 as sorbent and Cu/Ce0.6Zr0.4O2 as catalyst is reported by Hu et al. [165]. Four catalyst/sorbent layered configurations were investigated: mode I with one layered configuration (2 g/2 g), mode II and III with two layered configurations (2 g/2 g–2g/2g and 2 g/2 g–0.5g/0.5g) and mode IV (2 g/2 g–0.5g/0.5g–0.125g/0.125g), catalyst/sorbent, respectively. The optimum condition was a reaction temperature of 300 °C, a total pressure of 12 atm and an initial H2O/CO molar ratio of 1.5 with a three catalyst/sorbent layered configuration. This optimum condition yielded a H2 purity as high as 99.4% (dry basis) at the first SEWGS cycle, which was stabilized at 98.2% after 10 consecutive cycles, demonstrating good cyclic stability. Recently, the same authors [183] prepared a hybrid material, K2CO3-promoted Cu/MgO–Al2O3 by a sol–gel method. It was observed that the K/(Mg + Al) and Mg/Al atomic ratios affect the physicochemical properties of hybrid materials, especially in the morphology and the basicity distribution, which in turn affected the CO2 adsorption performance. In addition, it was found that the regeneration temperature of hybrid materials influences the SEWGS performance, 380 °C being the most favorable temperature since at higher temperatures the CO conversion at the post-breakthrough stage decreases with the number of cycles, but it does not happen for the material regenerated at 350 or 380 °C. The best performance was obtained for the hybrid material composed by a K/(Mg + Al) ratio of 0.2 and a Mg/Al ratio of 9, since the CO was completely converted and a yield >99.9% of H2 was attained in 10 consecutive SEWGS cycles at 300 °C, and regeneration at 380 °C.
The performance of hybrid materials is not consensual, further studies are needed. In relation to the sequential arrangement, both studies show an enhanced performance of SEWGS process with the increase in catalyst–sorbent layers. As mentioned above, also for sorbents at medium temperature, the preparation of catalysts and sorbents using wastes as precursors should be evaluated. In case of Mg-based materials, the potential of Mg recovered from magnesite mines sludges or from desalination reject brine [188] is an interesting alternative.
4. Conclusions and Recommendations
The goal of making Europe climate neutral in 2050 brings new challenges for the energy sector. Indeed, the decarbonization of power and heat, transports and stationary applications must be accelerated, and the use of hydrogen as an energy vector is considered the most promising alternative as, when burned, no CO2 is emitted to the atmosphere. Hydrogen does not exist naturally, and the most common processes for hydrogen production, i.e., steam methane reforming and coal gasification, produce large amounts of CO2 (>9 kg CO2/kg H2).
Considering hydrogen production using renewable energy sources, there are various technological options available with different TRLs and costs. The use of hydrogen as a storage means is also relevant, namely, when electricity is produced to overpass the power intermittency when using renewables’ sources. In the transition period to low carbon technologies, carbon capture and storage has a crucial role in the decarbonization and may offer a path to carbon neutrality in the context of a circular economy. Herein, in this paper the research progress of sorption-enhanced processes by in situ CO2 capture using solid sorbents during hydrogen production by reforming or gasification processes was reviewed. Syngas production (mainly CO, H2 and CO2) by these processes is performed at high temperatures (>600 °C), so it requires high-temperature CO2 sorbents such as Ca-based or alkali-based materials. On the other hand, medium-temperature sorbents (200–400 °C) such as Mg-based materials are used during the syngas upgrade through the water–gas shift reaction. The hydrogen production yield and purity will depend on sorbents’ CO2 carrying capacity and stability, and these properties are a target of study in several publications. However, most of the research is focused only on the sorbent point of view. On the contrary, the objective of this work was to assess how the sorbent performance affects the hydrogen production. The water–gas shift reaction is particularly relevant in this process as it acts as a separation technique and leads to the production of additional hydrogen, increasing the calorific value of syngas.
Among the sorbents used at high temperature, Ca-based ones appear to be the most promising due to the fast carbonation kinetics and high theoretical CO2 carrying capacity. However, deactivation along the time mainly due to sintering and pores blockage is still a limitation. Some approaches have been applied to improve their performance (e.g., use of inert additives, synthesis of nanomaterials or mixed oxides), but these strategies increase the cost of the sorbents. Alkali-based sorbents are more expensive than Ca-based ones, their carbonation is kinetically limited and the CO2 carrying capacity is lower. The doping with alkali molten salts appears to be the most promising option to increase sorbents’ reactivity, but the performance of these sorbents must be considerably improved to become competitive, at least when comparing with Ca-based sorbents. The regeneration of high-temperature sorbents, especially in the case of Ca-based ones which require temperatures above 900 ⁰C, increases the energy consumption of the process. Then, further efforts are required to reduce this gap, for instance, by investing in the implementation of energetic integration processes, using renewable sources during the calcination (e.g., solar reactors) or integrating chemical looping processes.
At a medium temperature, Mg-based sorbents are very interesting due to the highest theoretical CO2 carrying capacity, and because their carbonation temperature matches with the optimal conditions of water–gas shift reaction (250–400 °C). However, this sorbent presents slow kinetics, which is delaying its implementation as a CO2 sorbent. In the last years, the use of alkali molten salts allowed for considerably improving their performance, and CO2 carrying capacity values close to 0.6 g CO2/g sorbent are described in the literature. Thus, the next step should be focused on the improvement of Mg-based sorbents’ stability along the cycles.
The in situ CO2 capture requires sorbents with adequate activity under the operating conditions of reforming and gasification processes, or water–gas shift reaction. So, considering the operating conditions of these processes, the effects of temperature, steam and high-pressure on the sorbent’s performance were evaluated. In addition, the catalyst-sorbent synergies, using hybrid/mixed materials or sequential arrangement, was reviewed. The advantages of hybrid materials, such as sorbent’s ability to rapidly capture the CO2 generated during the process, and their drawbacks, such as the reduction in catalysts or sorbents’ active sites availability, are described in the literature, but the conclusions are often contradictory, and must be better understood.
For all the sorbents mentioned above, the regeneration step is usually performed under N2 or air atmosphere, which is not interesting for practical applications. Future studies should evaluate the effects of using high CO2 concentrated streams during the regeneration on sorbents’ performance and stability. In addition, the influence of impurities (e.g., sulphur) on the sorbents’ performance must be investigated. The combination of Ca-based and Mg-based materials can contribute to the sorbents CO2 capture performance. On one hand, the addition of magnesium materials to the Ca-based sorbents stabilizes the sorbents’ pore structure due to the high Tammann temperature of MgO, and hinders the CaO aggregation during Ca-looping. On the other hand, the addition of calcium materials to the Mg-based sorbents allows for a faster nucleation and growth of Mg-carbonates at medium-temperature sorption. Thus, the synergies between both precursors and the optimization of Ca/Mg ratios for high and medium sorption processes should be explored. For large-scale implementation, especially if fluidized bed reactors are used, granulation is sometimes the most suitable way to reduce the elutriation processes. Then, granulation processes should be further developed, and the mechanical properties of the materials should be assessed.
Ongoing research studies in sorption-enhanced processes are quite vast and should be a target of further research in the next decade. The performance of CO2 sorbents at high and medium temperatures should dictate the hydrogen production yield when reforming and gasification processes are used. The use of biomass-derived feedstocks is desirable, but during the energy transition, where the fossil fuels are still used, the in situ CO2 capture can simultaneously contribute to the improvement of the hydrogen yield production and the reduction in CO2 emissions.
To accelerate the large-scale implementation of the sorption-enhanced reforming or gasification processes, or sorption-enhanced water–gas shift reaction for hydrogen production with in situ CO2 capture using solid sorbents at high and medium temperature, respectively, future research studies should focus on the:
- Sorbents’ regeneration under realistic conditions, i.e., CO2 concentration higher than 70%.
- Sorbents obtained or synthetized using waste resources (e.g., CaO from paper and pulp industry sludges or CaO-rich biomass ashes; lithium from ores, brine, sea water or recycled batteries; MgO recovered from magnesite mine sludges or from desalination reject brine).
- Sorbents or sorbents–catalyst stability when a high number of carbonation–calcination cycles are performed (>50 carbonation–calcination cycles).
- Effect of granulation methodologies on sorbents or sorbents–catalyst reactivity and mechanical properties. The act of pressing and binders used during the pellet’s preparation can affect the porosity and reduce the catalysts–sorbents specific surface area. Then, the use of materials that can improve the materials porosity should be evaluated (e.g., biomass templates, ethylene glycol).
- Synergies between the hybrid, mixed or sequential arrangement of sorbents–catalyst. Since the carbonation of sorbents can block the access to the catalyst’s active sites, especially in the case of hybrid materials, the use of support materials or the increase in materials porosity that can reduce the occurrence of these problems should be evaluated.
- Tecno-economic viability of reforming, gasification and water–gas shift processes, considering, for example, the cost of the raw material, operating costs, energy requirements, and retrofit of existent industrial plants.
- Life cycle assessment of all the processes and the fulfilment of the circular economy concept. The use of different wastes as raw matter for the sorbents or catalyst synthesis should be compared.
- Modelling and numerical simulation of reforming, gasification and water–gas shift reactors for different catalyst/sorbent formulations foreseeing the processes’ upscale potential.
Finally, performing experiments on a pilot scale is essential to reaching a conclusion about the sorption-enhanced processes’ viability during the hydrogen production.
Author Contributions
P.T.: conceptualization, investigation, writing—original draft preparation, writing—review and editing; C.B.: writing—review and editing, P.C.: investigation, C.I.C.P.: writing—review and editing; I.C.: writing—review and editing, supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundação para a Ciência e Tecnologia (FCT) through projects UIDB/00100/2020 and UIDP/00100/2020 (Centro de Química Estrutural Research Unit), projects UIDB/04028/2020 and UIDP/04028/2020 (CERENA Research Unit), project LA/P/0056/2020 (Institute of Molecular Sciences Associate Laboratory) and Solar-driven Ca-Looping Process for Thermochemical Energy Storage (PTDC/EAM-PEC/32342/2017). Carmen Bacariza also thanks FCT for the contract 2020.00030.CEECIND.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMS | Alkali molten salts |
| CaL | Calcium looping |
| CCS | Carbon capture and storage |
| CCU | Carbon capture and utilization |
| CHL | Chemical looping hydrogen |
| CP | Conventional process |
| HTlc | Hydrotalcite-like-compound |
| HTS | High-temperature shift |
| IGCC | Integrated gasification combined cycles |
| LTS | Low-temperature shift |
| S/C | Steam/carbon |
| SE | Sorption enhanced |
| SEG | Sorption-enhanced gasification |
| SEP | Sorption-enhanced process |
| SER | Sorption-enhanced reforming |
| SESG | Sorption-enhanced steam gasification |
| SESR | Sorption-enhanced steam reforming |
| SEWGS | Sorption-enhanced water–gas shift |
| SMR | Steam methane reforming |
| TRL | Technology readiness level |
| WGS | Water–gas shift |
References
- European Commission. COM(2019) 640 final. Eur. Green Deal 2019, 53, 1689–1699. [Google Scholar] [CrossRef]
- Parkinson, B.; Balcombe, P.; Speirs, J.F.; Hawkes, A.D.; Hellgardt, K. Levelized cost of CO2 mitigation from hydrogen production routes. Energy Environ. Sci. 2019, 12, 19–40. [Google Scholar] [CrossRef]
- Thomas, H.; Armstrong, F.; Brandon, N.; David, B.; Barron, A.; Durrant, J.; Guwy, A.; Kucernak, A.; Lewis, M.; Maddy, J.; et al. Options for Producing Low-Carbon Hydrogen at Scale; Royal Society: London, UK, 2018; ISBN 9781782523185. [Google Scholar]
- Wang, Y.; Memon, M.Z.; Seelro, M.A.; Fu, W.; Gao, Y.; Dong, Y.; Ji, G. A review of CO2 sorbents for promoting hydrogen production in the sorption-enhanced steam reforming process. Int. J. Hydrogen Energy 2021, 46, 23358–23379. [Google Scholar] [CrossRef]
- Dou, B.; Zhang, H.; Song, Y.; Zhao, L.; Jiang, B.; He, M.; Ruan, C.; Chen, H.; Xu, Y. Hydrogen production from the thermochemical conversion of biomass: Issues and challenges. Sustain. Energy Fuels 2019, 3, 314–342. [Google Scholar] [CrossRef]
- Hu, Y.; He, Q.; Xu, C. Catalytic conversion of glycerol into hydrogen and value-added chemicals: Recent research advances. Catalysts 2021, 11, 1455. [Google Scholar] [CrossRef]
- Dang, C.; Yu, H.; Wang, H.; Peng, F.; Yang, Y. A bi-functional Co-CaO-Ca12Al14O33 catalyst for sorption-enhanced steam reforming of glycerol to high-purity hydrogen. Chem. Eng. J. 2016, 286, 329–338. [Google Scholar] [CrossRef]
- Macedo, M.S.; Soria, M.A.; Madeira, L.M. Process intensification for hydrogen production through glycerol steam reforming. Renew. Sustain. Energy Rev. 2021, 146, 111151. [Google Scholar] [CrossRef]
- Alves, H.J.; Bley Junior, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araújo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrogen Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Wu, X.; Wu, S. Production of high-purity hydrogen by sorption-enhanced steam reforming process of methanol. J. Energy Chem. 2015, 24, 315–321. [Google Scholar] [CrossRef]
- Aceves Olivas, D.Y.; Baray Guerrero, M.R.; Escobedo Bretado, M.A.; Marques Da Silva Paula, M.; Salinas Gutiérrez, J.; Guzmán Velderrain, V.; López Ortiz, A.; Collins-Martínez, V. Enhanced ethanol steam reforming by CO2 absorption using CaO, CaO∗MgO or Na2ZrO3. Int. J. Hydrogen Energy 2014, 39, 16595–16607. [Google Scholar] [CrossRef]
- Dewoolkar, K.D.; Vaidya, P.D. Tailored hydrotalcite-based hybrid materials for hydrogen production via sorption-enhanced steam reforming of ethanol. Int. J. Hydrogen Energy 2016, 41, 6094–6106. [Google Scholar] [CrossRef]
- Chimpae, S.; Wongsakulphasatch, S.; Vivanpatarakij, S. Syngas production from combined steam gasification of biochar and a sorption-enhanced. Processes 2019, 7, 349. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zheng, H.; Zheng, Y.; Tian, J.; Jing, T.; Chang, J.S.; Ho, S.H. Recent advances in hydrogen production by thermo-catalytic conversion of biomass. Int. J. Hydrogen Energy 2019, 44, 14266–14278. [Google Scholar] [CrossRef]
- Yan, J. Negative-emissions hydrogen energy. Nat. Clim. Chang. 2018, 8, 560–561. [Google Scholar] [CrossRef]
- Nazir, H.; Louis, C.; Jose, S.; Prakash, J.; Muthuswamy, N.; Buan, M.E.M.; Flox, C.; Chavan, S.; Shi, X.; Kauranen, P.; et al. Is the H2 economy realizable in the foreseeable future? Part I: H2 production methods. Int. J. Hydrogen Energy 2020, 45, 13777–13788. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Efstathiou, A.M. Hydrogen production technologies: Current state and future developments. Conf. Pap. Energy 2013, 2013, 690627. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, G.; Sun, Z.; Zhang, Y.; Xu, D. Review of renewable energy-based hydrogen production processes for sustainable energy innovation. Glob. Energy Interconnect. 2019, 2, 436–443. [Google Scholar] [CrossRef]
- Maggio, G.; Nicita, A.; Squadrito, G. How the hydrogen production from RES could change energy and fuel markets: A review of recent literature. Int. J. Hydrogen Energy 2019, 44, 11371–11384. [Google Scholar] [CrossRef]
- Baykara, S.Z. Hydrogen: A brief overview on its sources, production and environmental impact. Int. J. Hydrogen Energy 2018, 43, 10605–10614. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Voldsund, M.; Jordal, K.; Anantharaman, R. Hydrogen production with CO2 capture. Int. J. Hydrogen Energy 2016, 41, 4969–4992. [Google Scholar] [CrossRef]
- Cantuarias-Villessuzanne, C.; Weinberger, B.; Roses, L.; Vignes, A.; Brignon, J.M. Social cost-benefit analysis of hydrogen mobility in Europe. Int. J. Hydrogen Energy 2016, 41, 19304–19311. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Miller, J.E. Initial Case for Splitting Carbon Dioxide to Carbon Monoxide and Oxygen; Sandia National Laboratories: Albuquerque, NM, USA, 2007. [Google Scholar]
- Sun, H.; Wu, C.; Shen, B.; Zhang, X.; Huang, J.; Zhang, Y. Progress in the development and application of CaO-based adsorbents for CO2 capture—A review. Mater. Today Sustain. 2018, 1–2, 1–27. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, Y.; Sun, J.; Li, H.; Liu, W. Progress in MgO sorbents for cyclic CO2 capture: A comprehensive review. J. Mater. Chem. A 2019, 7, 20103–20120. [Google Scholar] [CrossRef]
- Yan, X.; Li, Y.; Ma, X.; Zhao, J.; Wang, Z. Performance of Li4SiO4 material for CO2 capture: A review. Int. J. Mol. Sci. 2019, 20, 928. [Google Scholar] [CrossRef] [Green Version]
- Dunstan, M.T.; Donat, F.; Bork, A.H.; Grey, C.P.; Müller, C.R. CO2 capture at medium to high temperature using solid oxide-based sorbents: Fundamental aspects, mechanistic insights, and recent advances. Chem. Rev. 2021, 121, 12681–12745. [Google Scholar] [CrossRef]
- Dubinin, A.M.; Tuponogov, V.G.; Ikonnikov, I.S. Modeling the process of producing hydrogen from methane. Theor. Found. Chem. Eng. 2013, 47, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Wang, T. Investigation of low rank coal gasification in a two-stage downdraft entrained-flow gasifier. Int. J. Clean Coal Energy 2014, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raibhole, V.N.; Sapali, S.N. Simulation and parametric analysis of cryogenic oxygen plant for biomass gasification. Mech. Eng. Res. 2012, 2, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Plis, P.; Wilk, R.K. Theoretical and experimental investigation of biomass gasification process in a fixed bed gasifier. Energy 2011, 36, 3838–3845. [Google Scholar] [CrossRef]
- Chen, L.; Qi, Z.; Zhang, S.; Su, J.; Somorjai, G.A. Catalytic hydrogen production from methane: A review on recent progress and prospect. Catalysts 2020, 10, 858. [Google Scholar] [CrossRef]
- Capa, A.; García, R.; Chen, D.; Rubiera, F.; Pevida, C.; Gil, M.V. On the effect of biogas composition on the H2 production by sorption enhanced steam reforming (SESR). Renew. Energy 2020, 160, 575–583. [Google Scholar] [CrossRef]
- Tomishige, K.; Asadullah, M.; Kunimori, K. Syngas production by biomass gasification using Rh/CeO2/SiO2 catalysts and fluidized bed reactor. Catal. Today 2004, 89, 389–403. [Google Scholar] [CrossRef]
- Antzaras, A.N.; Lemonidou, A.A. Recent advances on materials and processes for intensified production of blue hydrogen. Renew. Sustain. Energy Rev. 2021, 155, 111917. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Huang, X.; Feng, T.; Mu, M. Sorption-enhanced reaction process using advanced Ca-based sorbents for low-carbon hydrogen production. Process Saf. Environ. Prot. 2021, 155, 325–342. [Google Scholar] [CrossRef]
- Chisalita, D.A.; Cormos, C.C. Techno-economic assessment of hydrogen production processes based on various natural gas chemical looping systems with carbon capture. Energy 2019, 181, 331–344. [Google Scholar] [CrossRef]
- Kumar, G.; Eswari, A.P.; Kavitha, S.; Kumar, M.D.; Kannah, R.Y.; How, L.C.; Muthukaruppan, G.; Banu, J.R. Thermochemical conversion routes of hydrogen production from organic biomass: Processes, challenges and limitations. Biomass Convers. Biorefinery 2020. [Google Scholar] [CrossRef]
- Doranehgard, M.H.; Samadyar, H.; Mesbah, M.; Haratipour, P.; Samiezade, S. High-purity hydrogen production with in situ CO2 capture based on biomass gasification. Fuel 2017, 202, 29–35. [Google Scholar] [CrossRef]
- Parvez, A.M.; Hafner, S.; Hornberger, M.; Schmid, M.; Scheffknecht, G. Sorption enhanced gasification (SEG) of biomass for tailored syngas production with in-situ CO2 capture: Current status, process scale-up experiences and outlook. Renew. Sustain. Energy Rev. 2021, 141, 110756. [Google Scholar] [CrossRef]
- Mongkolsiri, P.; Jitkeaw, S.; Patcharavorachot, Y.; Arpornwichanop, A.; Assabumrungrat, S.; Authayanun, S. Comparative analysis of biomass and coal based co-gasification processes with and without CO2 capture for HT-PEMFCs. Int. J. Hydrogen Energy 2019, 44, 2216–2229. [Google Scholar] [CrossRef]
- Fermoso, J.; Rubiera, F.; Chen, D. Sorption enhanced catalytic steam gasification process: A direct route from lignocellulosic biomass to high purity hydrogen. Energy Environ. Sci. 2012, 5, 6358–6367. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Gilot, P. Review-calcination and carbonation of limestone during thermal cycling for CO2 sequestration. Fuel Process. Technol. 2005, 86, 1707–1743. [Google Scholar] [CrossRef]
- Halliday, C.; Hatton, T.A. Sorbents for the capture of CO2 and other acid gases: A review. Ind. Eng. Chem. Res. 2021, 60, 9313–9346. [Google Scholar] [CrossRef]
- Radfarnia, H.R.; Iliuta, M.C. Metal oxide-stabilized calcium oxide CO2 sorbent for multicycle operation. Chem. Eng. J. 2013, 232, 280–289. [Google Scholar] [CrossRef]
- Chi, C.; Li, Y.; Zhang, W.; Wang, Z. Synthesis of a hollow microtubular Ca/Al sorbent with high CO2 uptake by hard templating. Appl. Energy 2019, 251, 113382. [Google Scholar] [CrossRef]
- Skoufa, Z.; Antzara, A.; Heracleous, E.; Lemonidou, A.A. Evaluating the activity and stability of CaO-based sorbents for post-combustion CO2 capture in fixed-bed reactor experiments. Energy Procedia 2016, 86, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Drese, J.H.; Jones, C.W. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem 2009, 2, 796–854. [Google Scholar] [CrossRef]
- Teixeira, P.; Mohamed, I.; Fernandes, A.; Silva, J.; Ribeiro, F.; Pinheiro, C.I.C. Enhancement of sintering resistance of CaO-based sorbents using industrial waste resources for Ca-looping in the cement industry. Sep. Purif. Technol. 2020, 235, 116190. [Google Scholar] [CrossRef]
- Peng, W.; Xu, Z.; Zhao, H. Batch fluidized bed test of SATS-derived CaO/TiO2–Al2O3 sorbent for calcium looping. Fuel 2016, 170, 226–234. [Google Scholar] [CrossRef]
- Valverde, J.M.; Medina, S. Crystallographic transformation of limestone during calcination under CO2. Phys. Chem. Chem. Phys. 2015, 17, 21912–21926. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Feng, Y.; Liu, L.; Guo, X. Effects of preparation methods on the structure and property of Al-stabilized CaO-based sorbents for CO2 capture. Fuel Process. Technol. 2018, 173, 276–284. [Google Scholar] [CrossRef]
- Valverde, J.M.; Sanchez-Jimenez, P.E.; Perez-Maqueda, L.A. Ca-looping for postcombustion CO2 capture: A comparative analysis on the performances of dolomite and limestone. Appl. Energy 2015, 138, 202–215. [Google Scholar] [CrossRef] [Green Version]
- Arstad, B.B.; Lind, A.; Andreassen, K.A.; Pierchala, J.; Thorshaug, K.; Blom, R. In-situ XRD studies of dolomite based CO2 sorbents. Energy Procedia 2014, 63, 2082–2091. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.C.; Phalak, N.; Sun, Z.; Fan, L.S. Activation strategies for calcium-based sorbents for CO2 capture: A perspective. Ind. Eng. Chem. Res. 2012, 51, 2133–2142. [Google Scholar] [CrossRef]
- Santos, E.T.; Alfonsín, C.; Chambel, A.J.S.; Fernandes, A.; Soares Dias, A.P.; Pinheiro, C.I.C.; Ribeiro, M.F. Investigation of a stable synthetic sol-gel CaO sorbent for CO2 capture. Fuel 2012, 94, 624–628. [Google Scholar] [CrossRef]
- Teixeira, P.; Hipólito, J.; Fernandes, A.; Ribeiro, F.; Pinheiro, C.I.C. Tailoring synthetic gol-gel CaO sorbents with high reactivity or high stability for Ca-Looping CO2 Capture. Ind. Eng. Chem. Res. 2019, 58, 8484–8494. [Google Scholar] [CrossRef]
- Pinheiro, C.I.C.; Fernandes, A.; Freitas, C.; Santos, E.T.; Ribeiro, M.F. Waste Marble Powders as Promising Inexpensive Natural CaO-Based Sorbents for Post-Combustion CO2 Capture. Ind. Eng. Chem. Res. 2016, 55, 7860–7872. [Google Scholar] [CrossRef]
- Nawar, A.; Ghaedi, H.; Ali, M.; Zhao, M.; Iqbal, N.; Khan, R. Recycling waste-derived marble powder for CO2 capture. Process Saf. Environ. Prot. 2019, 132, 214–225. [Google Scholar] [CrossRef]
- He, S.; Hu, Y.; Hu, T.; Ma, A.; Jia, Q.; Su, H.; Shan, S. Investigation of CaO-based sorbents derived from eggshells and red mud for CO2 capture. J. Alloys Compd. 2017, 701, 828–833. [Google Scholar] [CrossRef]
- Li, Y.; Sun, R. Studies on adsorption of carbon dioxide on alkaline paper mill waste using cyclic process. Energy Convers. Manag. 2014, 82, 46–53. [Google Scholar] [CrossRef]
- Nawar, A.; Ali, M.; Mahmood, M.; Anwar, M.; Khan, Z.A. Effect of structural promoters on calcium based sorbents from waste derived sources. Mater. Today Commun. 2020, 24, 101075. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Sun, R.; Wu, S.; Lu, C. Thermal analysis of cyclic carbonation behavior of CaO derived from carbide slag at high temperature. J. Therm. Anal. Calorim. 2012, 110, 685–694. [Google Scholar] [CrossRef]
- Chi, C.; Li, Y.; Ma, X.; Duan, L. CO2 capture performance of CaO modified with by-product of biodiesel at calcium looping conditions. Chem. Eng. J. 2017, 326, 378–388. [Google Scholar] [CrossRef]
- Yasipourtehrani, S.; Tian, S.; Strezov, V.; Kan, T.; Evans, T. Development of robust CaO-based sorbents from blast furnace slag for calcium looping CO2 capture. Chem. Eng. J. 2020, 387, 124140. [Google Scholar] [CrossRef]
- Shan, S.Y.; Ma, A.H.; Hu, Y.C.; Jia, Q.M.; Wang, Y.M.; Peng, J.H. Development of sintering-resistant CaO-based sorbent derived from eggshells and bauxite tailings for cyclic CO2 capture. Environ. Pollut. 2015, 208, 546–552. [Google Scholar] [CrossRef]
- Witoon, T. Characterization of calcium oxide derived from waste eggshell and its application as CO2 sorbent. Ceram. Int. 2011, 37, 3291–3298. [Google Scholar] [CrossRef]
- Castilho, S.; Kiennemann, A.; Costa Pereira, M.F.; Soares Dias, A.P. Sorbents for CO2 capture from biogenesis calcium wastes. Chem. Eng. J. 2013, 226, 146–153. [Google Scholar] [CrossRef]
- Salaudeen, S.A.; Tasnim, S.H.; Heidari, M.; Acharya, B.; Dutta, A. Eggshell as a potential CO2 sorbent in the calcium looping gasification of biomass. Waste Manag. 2018, 80, 274–284. [Google Scholar] [CrossRef]
- Yan, F.; Jiang, J.; Li, K.; Liu, N.; Chen, X.; Gao, Y.; Tian, S. Green synthesis of nanosilica from coal fly ash and its stabilizing effect on CaO sorbents for CO2 capture. Environ. Sci. Technol. 2017, 51, 7606–7615. [Google Scholar] [CrossRef]
- Nawar, A.; Ali, M.; Waqas, A.; Javed, A.; Iqbal, N.; Khan, R.; Ash, F. Effect of different activation processes on CaO/fly ash mixture for CO2 capture. Energy Fuels 2020, 34, 2035–2044. [Google Scholar] [CrossRef]
- Mohamed, M.; Yusup, S.; Bustam, M.A.; Azmi, N. Effect of coal bottom ash and binder addition into CaO-based sorbent on CO2 capture performance. Chem. Eng. Trans. 2017, 56, 325–330. [Google Scholar] [CrossRef]
- Yan, F.; Jiang, J.; Li, K.; Tian, S.; Zhao, M.; Chen, X. Performance of coal fly ash stabilized, CaO-based sorbents under different carbonation-calcination conditions. ACS Sustain. Chem. Eng. 2015, 3, 2092–2099. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Ren, Q.; Duan, L.; Chen, H.; Chen, X. Effect of rice husk ash addition on CO2 capture behavior of calcium-based sorbent during calcium looping cycle. Fuel Process. Technol. 2009, 90, 825–834. [Google Scholar] [CrossRef]
- Hu, Y.C.; Liu, W.Q.; Yang, Y.D.; Sun, J.; Zhou, Z.J.; Xu, M.H. Enhanced CO2 capture performance of limestone by industrial waste sludge. Chem. Eng. Technol. 2017, 40, 2322–2328. [Google Scholar] [CrossRef]
- Su, C.; Duan, L.; Donat, F.; Anthony, E.J. From waste to high value utilization of spent bleaching clay in synthesizing high-performance calcium-based sorbent for CO2 capture. Appl. Energy 2018, 210, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Salaudeen, S.A.; Acharya, B.; Dutta, A. CaO-based CO2 sorbents: A review on screening, enhancement, cyclic stability, regeneration and kinetics modelling. J. CO2 Util. 2018, 23, 179–199. [Google Scholar] [CrossRef]
- Esence, T.; Guillot, E.; Tessonneaud, M.; Sans, J.L.; Flamant, G. Solar calcination at pilot scale in a continuous flow multistage horizontal fluidized bed. Sol. Energy 2020, 207, 367–378. [Google Scholar] [CrossRef]
- Tregambi, C.; Troiano, M.; Montagnaro, F.; Solimene, R.; Salatino, P. Fluidized beds for concentrated solar thermal technologies—A review. Front. Energy Res. 2021, 9, 618421. [Google Scholar] [CrossRef]
- Rivero, M.A.; Rodrigues, D.; Pinheiro, C.I.C.; Cardoso, J.P.; Mendes, L.F. Solid – gas reactors driven by concentrated solar energy with potential application to calcium looping: A comparative review. Renew. Sustain. Energy Rev. 2022, 158, 112048. [Google Scholar] [CrossRef]
- Dang, C.; Wu, S.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Co-production of high quality hydrogen and synthesis gas via sorption-enhanced steam reforming of glycerol coupled with methane reforming of carbonates. Chem. Eng. J. 2019, 360, 47–53. [Google Scholar] [CrossRef]
- Dang, C.; Long, J.; Li, H.; Cai, W.; Yu, H. Pd-promoted Ni-Ca-Al bi-functional catalyst for integrated sorption-enhanced steam reforming of glycerol and methane reforming of carbonate. Chem. Eng. Sci. 2021, 230, 116226. [Google Scholar] [CrossRef]
- Mahishi, M.R.; Goswami, D.Y. An experimental study of hydrogen production by gasification of biomass in the presence of a CO2 sorbent. Int. J. Hydrogen Energy 2007, 32, 2803–2808. [Google Scholar] [CrossRef]
- Irfan, M.; Li, A.; Zhang, L.; Wang, M.; Chen, C.; Khushk, S. Production of hydrogen enriched syngas from municipal solid waste gasification with waste marble powder as a catalyst. Int. J. Hydrogen Energy 2019, 44, 8051–8061. [Google Scholar] [CrossRef]
- Irfan, M.; Li, A.; Zhang, L.; Javid, M.; Wang, M.; Khushk, S. Enhanced H2 production from municipal solid waste gasification using Ni-CaO-TiO2 bifunctional catalyst prepared by dc arc plasma melting. Ind. Eng. Chem. Res. 2019, 58, 13408–13419. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, C.; Ying, Z.; Wang, B. Experimental study on gasification performance of bamboo and PE from municipal solid waste in a bench-scale fixed bed reactor. Energy Convers. Manag. 2016, 117, 393–399. [Google Scholar] [CrossRef]
- Wang, J.; Kang, D.; Shen, B.; Sun, H.; Wu, C. Enhanced hydrogen production from catalytic biomass gasification with in-situ CO2 capture. Environ. Pollut. 2020, 267, 115487. [Google Scholar] [CrossRef]
- Shahbaz, M.; Yusup, S.; Inayat, A.; Patrick, D.O.; Ammar, M.; Pratama, A. Cleaner production of hydrogen and syngas from catalytic steam palm kernel shell gasification using CaO sorbent and coal bottom ash as a catalyst. Energy Fuels 2017, 31, 13824–13833. [Google Scholar] [CrossRef]
- Arnold, R.A.; Hill, J.M. Catalysts for gasification: A review. Sustain. Energy Fuels 2019, 3, 656–672. [Google Scholar] [CrossRef]
- Detchusananard, T.; Im-orb, K.; Ponpesh, P.; Arpornwichanop, A. Biomass gasification integrated with CO2 capture processes for high-purity hydrogen production: Process performance and energy analysis. Energy Convers. Manag. 2018, 171, 1560–1572. [Google Scholar] [CrossRef]
- Hanak, D.P.; Michalski, S.; Manovic, V. From post-combustion carbon capture to sorption-enhanced hydrogen production: A state-of-the-art review of carbonate looping process feasibility. Energy Convers. Manag. 2018, 177, 428–452. [Google Scholar] [CrossRef]
- Rosa, L.G. Solar heat for materials processing: A review on recent achievements and a prospect on future trends. ChemEngineering 2019, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Fuchs, J.; Schmid, J.C.; Benedikt, F.; Hofbauer, H. Experimental development of sorption enhanced reforming by the use of an advanced gasification test plant. Int. J. Hydrogen Energy 2017, 42, 29694–29707. [Google Scholar] [CrossRef]
- Guoxin, H.; Hao, H. Hydrogen rich fuel gas production by gasification of wet biomass using a CO2 sorbent. Biomass Bioenergy 2009, 33, 899–906. [Google Scholar] [CrossRef]
- Criado, Y.A.; Arias, B.; Abanades, J.C. Effect of the carbonation temperature on the CO2 carrying capacity of CaO. Ind. Eng. Chem. Res. 2018, 57, 12595–12599. [Google Scholar] [CrossRef] [Green Version]
- Donat, F.; Florin, N.H.; Anthony, E.J.; Fennell, P.S. Influence of high-temperature steam on the reactivity of CaO sorbent for CO2 capture. Environ. Sci. Technol. 2012, 46, 1262–1269. [Google Scholar] [CrossRef]
- Dong, J.; Tang, Y.; Nzihou, A.; Weiss-Hortala, E. Effect of steam addition during carbonation, calcination or hydration on re-activation of CaO sorbent for CO2 capture. J. CO2 Util. 2020, 39, 101167. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Guo, X.; Van Duin, A.C.T. Insights into the Role of H2O in the Carbonation of CaO Nanoparticle with CO2. J. Phys. Chem. C 2018, 122, 21401–21410. [Google Scholar] [CrossRef]
- Dou, B.; Song, Y.; Liu, Y.; Feng, C. High temperature CO2 capture using calcium oxide sorbent in a fixed-bed reactor. J. Hazard. Mater. 2010, 183, 759–765. [Google Scholar] [CrossRef]
- Donat, F. The Influence of High-Temperature Steam on the Rate and Extent of Reaction of CaO-Sorbent for CO2 Capture-and-Release. Master’s Thesis, TU Bergakademie Freiberg, Freiberg, Germany, 2011. [Google Scholar]
- Champagne, S.; Lu, D.Y.; MacChi, A.; Symonds, R.T.; Anthony, E.J. Influence of steam injection during calcination on the reactivity of CaO-based sorbent for carbon capture. Ind. Eng. Chem. Res. 2013, 52, 2241–2246. [Google Scholar] [CrossRef]
- Champagne, S.; Lu, D.Y.; Symonds, R.T.; Macchi, A.; Anthony, E.J.J. The effect of steam addition to the calciner in a calcium looping pilot plant. Powder Technol. 2015, 290, 114–123. [Google Scholar] [CrossRef]
- Tan, R.S.; Tuan Abdullah, T.A.; Ripin, A.; Ahmad, A.; Md Isa, K. Hydrogen-rich gas production by steam reforming of gasified biomass tar over Ni/dolomite/La2O3 catalyst. J. Environ. Chem. Eng. 2019, 7, 103490. [Google Scholar] [CrossRef]
- Iulianelli, A.; Manzolini, G.; De Falco, M.; Campanari, S.; Longo, T.; Liguori, S.; Basile, A. H2 production by low pressure methane steam reforming in a Pd-Ag membrane reactor over a Ni-based catalyst: Experimental and modeling. Int. J. Hydrogen Energy 2010, 35, 11514–11524. [Google Scholar] [CrossRef]
- Butler, J.W.; Grace, J.R. High-Pressure Systems and Processes for Calcium Looping; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; ISBN 9780857097606. [Google Scholar]
- Yu, F.C.; Fan, L.S. Kinetic study of high-pressure carbonation reaction of calcium-based sorbents in the calcium looping process (CLP). Ind. Eng. Chem. Res. 2011, 50, 11528–11536. [Google Scholar] [CrossRef]
- Shahid, M.M.; Abbas, S.Z.; Maqbool, F.; Ramirez-Solis, S.; Dupont, V.; Mahmud, T. Modeling of sorption enhanced steam methane reforming in an adiabatic packed bed reactor using various CO2 sorbents. J. Environ. Chem. Eng. 2021, 9, 105863. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Cheng, Z.; Luo, X.; Jia, L.; Song, M.; Jiang, B.; Dou, B. Sorption-enhanced steam reforming of glycerol for hydrogen production over a NiO/NiAl2O4 Catalyst and Li2ZrO3-Based Sorbent. Energy Fuels 2015, 29, 7408–7418. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Lacalle-Vilà, C.; Zhao, T.; Rønning, M.; Chen, D. Experimental demonstration of H2 production by CO2 sorption enhanced steam methane reforming using ceramic acceptors. Stud. Surf. Sci. Catal. 2007, 167, 159–164. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, C.; Chen, Y.; Cai, X.; Dou, B.; Chen, H.; Xu, Y.; Jiang, B.; Wang, K. High purity hydrogen production from sorption enhanced chemical looping glycerol reforming: Application of NiO-based oxygen transfer materials and potassium promoted Li2ZrO3 as CO2 sorbent. Appl. Therm. Eng. 2017, 124, 454–465. [Google Scholar] [CrossRef]
- Yang, X.; Liu, W.; Sun, J.; Hu, Y.; Wang, W.; Chen, H.; Zhang, Y.; Li, X.; Xu, M. Preparation of Novel Li4SiO4 sorbents with superior performance at low CO2 Concentration. ChemSusChem 2016, 9, 1607–1613. [Google Scholar] [CrossRef]
- Chen, S.; Dai, J.; Qin, C.; Yuan, W.; Manovic, V. Adsorption and desorption equilibrium of Li4SiO4-based sorbents for high-temperature CO2 capture. Chem. Eng. J. 2022, 429, 132236. [Google Scholar] [CrossRef]
- Abanades, J.C.; Rubin, E.S.; Anthony, E.J. Sorbent cost and performance in CO2 capture systems. Ind. Eng. Chem. Res. 2004, 43, 3462–3466. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Hong, M.; Liu, S.; Zhang, Y.; Liu, H.; Gu, C. CO2 absorption properties of Ti- and Na-doped porous Li4SiO4 prepared by a sol–gel process. J. Mater. Sci. 2015, 50, 4698–4706. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, S.; Hu, Y.; Sun, J.; Cao, J.; Li, Q.; Liu, W. Mechanochemically activated Li4SiO4-based adsorbent with enhanced CO2 capture performance and its modification mechanisms. Fuel 2020, 273, 117749. [Google Scholar] [CrossRef]
- Subha, P.V.; Nair, B.N.; Visakh, V.; Achu, R.; Shikhila, S.; Peer Mohamed, A.; Yamaguchi, T.; Hareesh, U.S. Wet chemically derived Li4SiO4 nanowires as efficient CO2 sorbents at intermediate temperatures. Chem. Eng. J. 2021, 406, 126731. [Google Scholar] [CrossRef]
- Zhang, T.; Li, M.; Ning, P.; Jia, Q.; Wang, Q.; Wang, J. K2CO3 promoted novel Li4SiO4-based sorbents from sepiolite with high CO2 capture capacity under different CO2 partial pressures. Chem. Eng. J. 2020, 380, 122515. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, M.J.; Kwon, Y.M.; Chae, H.J.; Cho, M.S.; Park, Y.K.; Seo, H.M.; Kim, J.C. Novel regenerable solid sorbents based on lithium orthosilicate for carbon dioxide capture at high temperatures. Sep. Purif. Technol. 2019, 214, 120–127. [Google Scholar] [CrossRef]
- Li, H.; Qu, M.; Hu, Y. High-temperature CO2 capture by Li4SiO4 adsorbents: Effects of pyroligneous acid (PA) modification and existence of CO2 at desorption stage. Fuel Process. Technol. 2020, 197, 106186. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Pfeiffer, H.; Louis, B.; Sun, L.; O’Hare, D.; Wang, Q. Recent advances in lithium containing ceramic based sorbents for high-temperature CO2 capture. J. Mater. Chem. A 2019, 7, 7962–8005. [Google Scholar] [CrossRef]
- Radfarnia, H.R.; Iliuta, M.C. Surfactant-template/ultrasound-assisted method for the preparation of porous nanoparticle lithium zirconate. Ind. Eng. Chem. Res. 2011, 50, 9295–9305. [Google Scholar] [CrossRef]
- Martinez-Cruz, L.; Pfeiffer, H. Microstructural Thermal Evolution of the Na2CO3 Phase Produced during a Na2ZrO3−CO2 Chemisorption Process. J. Phys. Chem. 2012, 49, 9038–9042. [Google Scholar]
- Shokrollahi Yancheshmeh, M.; Radfarnia, H.R.; Iliuta, M.C. High temperature CO2 sorbents and their application for hydrogen production by sorption enhanced steam reforming process. Chem. Eng. J. 2016, 283, 420–444. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Haugen, G.; Zhao, T.; Rønning, M.; Aartun, I.; Børresen, B.; Rytter, E.; Rønnekleiv, M.; Chen, D. Process design simulation of H2 production by sorption enhanced steam methane reforming: Evaluation of potential CO2 acceptors. Green Chem. 2007, 9, 654–666. [Google Scholar] [CrossRef]
- Xiao, Q.; Tang, X.; Liu, Y.; Zhong, Y.; Zhu, W. Citrate route to prepare K-doped Li2ZrO3 sorbents with excellent CO2 capture properties. Chem. Eng. J. 2011, 174, 231–235. [Google Scholar] [CrossRef]
- Kwon, Y.M.; Lee, S.C.; Chae, H.J.; Cho, M.S.; Park, Y.K.; Seo, H.M.; Chang Kim, J. Regenerable sodium-based lithium silicate sorbents with a new mechanism for CO2 capture at high temperature. Renew. Energy 2019, 144, 180–187. [Google Scholar] [CrossRef]
- Essaki, K.; Muramatsu, T.; Kato, M. Effect of equilibrium shift by using lithium silicate pellets in methane steam reforming. Int. J. Hydrogen Energy 2008, 33, 4555–4559. [Google Scholar] [CrossRef]
- Sanna, A.; Thompson, S.; Whitty, K.J.; Maroto-Valer, M.M. Fly ash derived lithium silicate for in-situ pre-combustion CO2 capture. Energy Procedia 2017, 114, 2401–2404. [Google Scholar] [CrossRef]
- Xiong, R.; Ida, J.; Lin, Y.S. Kinetics of carbon dioxide sorption on potassium-doped lithium zirconate. Chem. Eng. Sci. 2003, 58, 4377–4385. [Google Scholar] [CrossRef]
- Park, Y.C.; Jo, S.-H.; Ryu, C.K.; Yi, C.-K. Demonstration of pilot scale carbon dioxide capture system using dry regenerable sorbents to the real coal-fired power plant in Korea. Energy Procedia 2011, 4, 1508–1512. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shen, C.; Zhang, S.; Wu, Y. Steam methane reforming reaction enhanced by a novel K2CO3-Doped Li4SiO4 sorbent: Investigations on the sorbent and catalyst coupling behaviors and sorbent regeneration strategy. Int. J. Hydrogen Energy 2016, 41, 4831–4842. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Zhao, T.; Rønning, M.; Chen, D. Effects of steam addition on the properties of high temperature ceramic CO2 acceptors. J. Environ. Eng. 2009, 135, 397–403. [Google Scholar] [CrossRef]
- Xie, M.; Zhou, Z.; Qi, Y.; Cheng, Z.; Yuan, W. Sorption-enhanced steam methane reforming by in situ CO2 capture on a CaO-Ca9Al6O18 sorbent. Chem. Eng. J. 2012, 207–208, 142–150. [Google Scholar] [CrossRef]
- Pecharaumporn, P.; Wongsakulphasatch, S.; Glinrun, T.; Maneedaeng, A.; Hassan, Z.; Assabumrungrat, S. Synthetic CaO-based sorbent for high-temperature CO2 capture in sorption-enhanced hydrogen production. Int. J. Hydrogen Energy 2019, 44, 20663–20677. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Hosseini, D.; Armutlulu, A.; Margossian, T.; Copéret, C.; Müller, C. Bi-functional Ru/Ca3Al2O6-CaO catalyst-CO2 sorbent for the production of high purity hydrogen: Via sorption-enhanced steam methane reforming. Catal. Sci. Technol. 2019, 9, 5745–5756. [Google Scholar] [CrossRef] [Green Version]
- Di Giuliano, A.; Girr, J.; Massacesi, R.; Gallucci, K.; Courson, C. Sorption enhanced steam methane reforming by Ni–CaO materials supported on mayenite. Int. J. Hydrogen Energy 2017, 42, 13661–13680. [Google Scholar] [CrossRef]
- Di Giuliano, A.; Gallucci, K.; Kazi, S.S.; Giancaterino, F.; Di Carlo, A.; Courson, C.; Meyer, J.; Di Felice, L. Development of Ni- and CaO-based mono- and bi-functional catalyst and sorbent materials for sorption enhanced steam methane reforming: Performance over 200 cycles and attrition tests. Fuel Process. Technol. 2019, 195, 106160. [Google Scholar] [CrossRef]
- Masoudi Soltani, S.; Lahiri, A.; Bahzad, H.; Clough, P.; Gorbounov, M.; Yan, Y. Sorption-enhanced steam methane reforming for combined CO2 capture and hydrogen production: A state-of-the-art review. Carbon Capture Sci. Technol. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Di Giuliano, A.; Giancaterino, F.; Courson, C.; Foscolo, P.U.; Gallucci, K. Development of a Ni-CaO-mayenite combined sorbent-catalyst material for multicycle sorption enhanced steam methane reforming. Fuel 2018, 234, 687–699. [Google Scholar] [CrossRef]
- Phromprasit, J.; Powell, J.; Wongsakulphasatch, S.; Kiatkittipong, W.; Bumroongsakulsawat, P.; Assabumrungrat, S. Activity and stability performance of multifunctional catalyst (Ni/CaO and Ni/Ca12Al14O33-CaO) for bio-hydrogen production from sorption enhanced biogas steam reforming. Int. J. Hydrogen Energy 2016, 41, 7318–7331. [Google Scholar] [CrossRef]
- Santos, D.B.L.; Oliveira, A.C.P.; Hori, C.E. Performance of Na2CO3-CaO sorbent in sorption-enhanced steam methane reforming. J. CO2 Util. 2021, 51, 101634. [Google Scholar] [CrossRef]
- Wu, Y.J.; Li, P.; Yu, J.G.; Cunha, A.F.; Rodrigues, A.E. Progress on sorption-enhanced reaction process for hydrogen production. Rev. Chem. Eng. 2016, 32, 271–303. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Chen, Y.; Guo, X. Lithium-based sorbent from rice husk materials for hydrogen production via sorption-enhanced steam reforming of ethanol. Fuel 2019, 245, 263–273. [Google Scholar] [CrossRef]
- Swain, B. Recovery and recycling of lithium: A review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Coman, V.; Robotin, B.; Ilea, P. Nickel recovery/removal from industrial wastes: A review. Resour. Conserv. Recycl. 2013, 73, 229–238. [Google Scholar] [CrossRef]
- Chen, C.H.; Yu, C.T.; Chen, W.H.; Kuo, H.T. Effect of in-situ carbon dioxide sorption on methane reforming by nickel-calcium composite catalyst for hydrogen production. IOP Conf. Ser. Earth Environ. Sci. 2020, 463, 012102. [Google Scholar] [CrossRef]
- Dewoolkar, K.D.; Vaidya, P.D. Improved hydrogen production by sorption-enhanced steam methane reforming over hydrotalcite- and calcium-based hybrid materials. Energy Fuels 2015, 29, 3870–3878. [Google Scholar] [CrossRef]
- Johnsen, K.; Ryu, H.J.; Grace, J.R.; Lim, C.J. Sorption-enhanced steam reforming of methane in a fluidized bed reactor with dolomite as CO2-acceptor. Chem. Eng. Sci. 2006, 61, 1195–1202. [Google Scholar] [CrossRef]
- Rahmanzadeh, L.; Taghizadeh, M. Sorption-enhanced ethanol steam reforming on Ce-Ni/MCM-41 with simultaneous CO2 adsorption over Na- and Zr- promoted CaO based sorbent. Int. J. Hydrogen Energy 2019, 44, 21238–21250. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Guo, X.; Ni, S. Continuous high-purity hydrogen production by sorption enhanced steam reforming of ethanol over modified lithium silicate. Int. J. Hydrogen Energy 2021, 46, 10119–10130. [Google Scholar] [CrossRef]
- Phromprasit, J.; Powell, J.; Wongsakulphasatch, S.; Kiatkittipong, W.; Bumroongsakulsawat, P.; Assabumrungrat, S. H2 production from sorption enhanced steam reforming of biogas using multifunctional catalysts of Ni over Zr-, Ce- and La-modified CaO sorbents. Chem. Eng. J. 2017, 313, 1415–1425. [Google Scholar] [CrossRef]
- Xie, H.; Yu, Q.; Zuo, Z.; Han, Z.; Yao, X.; Qin, Q. Hydrogen production via sorption-enhanced catalytic steam reforming of bio-oil. Int. J. Hydrogen Energy 2016, 41, 2345–2353. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, H.; Wei, L.; Shao, J.; Wang, X.; Chen, H. Absorption-enhanced steam gasification of biomass for hydrogen production: Effects of calcium-based absorbents and NiO-based catalysts on corn stalk pyrolysis-gasification. Int. J. Hydrogen Energy 2017, 42, 5840–5848. [Google Scholar] [CrossRef]
- Mostafavi, E.; Mahinpey, N.; Rahman, M.; Sedghkerdar, M.H.; Gupta, R. High-purity hydrogen production from ash-free coal by catalytic steam gasification integrated with dry-sorption CO2 capture. Fuel 2016, 178, 272–282. [Google Scholar] [CrossRef]
- Zamboni, I.; Debal, M.; Matt, M.; Girods, P.; Kiennemann, A.; Rogaume, Y.; Courson, C. Catalytic gasification of biomass (Miscanthus) enhanced by CO2 sorption. Environ. Sci. Pollut. Res. 2016, 23, 22253–22266. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Chen, C.Y. Water gas shift reaction for hydrogen production and carbon dioxide capture: A review. Appl. Energy 2020, 258, 114078. [Google Scholar] [CrossRef]
- Baraj, E.; Ciahotný, K.; Hlinčik, T.; Šnajdrová, V. Hydrogen production via water gas shift reaction on a nickel based catalyst. PALIVA 2016, 8, 138–142. [Google Scholar] [CrossRef]
- Newsome, D.S. The water-gas shift reaction. Catal. Rev. Technol. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Smith J, B.R.; Loganathan, M.; Shekhar Shantha, M. A Review of the water gas shift reaction kinetics. Int. J. Chem. React. Eng. 2010, 8, 49. [Google Scholar] [CrossRef]
- Jin, S.; Ko, K.J.; Lee, C.H. Direct formation of hierarchically porous MgO-based sorbent bead for enhanced CO2 capture at intermediate temperatures. Chem. Eng. J. 2019, 371, 64–77. [Google Scholar] [CrossRef]
- Oliveira, N.M.B.; Valença, G.P.; Vieira, R. Water gas shift reaction on copper catalysts supported on alumina and carbon nanofibers. Chem. Eng. Trans. 2015, 43, 931–936. [Google Scholar] [CrossRef]
- Hwang, B.W.; Lim, J.H.; Chae, H.J.; Ryu, H.J.; Lee, D.; Lee, J.B.; Kim, H.; Lee, S.C.; Kim, J.C. CO2 capture and regeneration properties of MgO-based sorbents promoted with alkali metal nitrates at high pressure for the sorption enhanced water gas shift process. Process Saf. Environ. Prot. 2018, 116, 219–227. [Google Scholar] [CrossRef]
- Hu, Y.; Cui, H.; Cheng, Z.; Zhou, Z. Sorption-enhanced water gas shift reaction by in situ CO2 capture on an alkali metal salt-promoted MgO-CaCO3 sorbent. Chem. Eng. J. 2019, 377, 119823. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, K.B. Sorption-enhanced water gas shift reaction for high-purity hydrogen production: Application of a Na-Mg double salt-based sorbent and the divided section packing concept. Appl. Energy 2017, 205, 316–322. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, T.; Gao, Y.; Wang, Q. Enhanced water gas shift processes for carbon dioxide capture and hydrogen production. Appl. Energy 2019, 254, 113700. [Google Scholar] [CrossRef]
- Soria, M.A.; Rocha, C.; Tosti, S.; Mendes, A.; Madeira, L.M. COx free hydrogen production through water-gas shift reaction in different hybrid multifunctional reactors. Chem. Eng. J. 2019, 356, 727–736. [Google Scholar] [CrossRef]
- Halabi, M.H.; De Croon, M.H.J.M.; Van Der Schaaf, J.; Cobden, P.D.; Schouten, J.C. High capacity potassium-promoted hydrotalcite for CO2 capture in H2 production. Int. J. Hydrogen Energy 2012, 37, 4516–4525. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, S.; Yoon, H.J.; Yoon, C.W.; Lee, K.B. Water gas shift and sorption-enhanced water gas shift reactions using hydrothermally synthesized novel Cu–Mg–Al hydrotalcite-based catalysts for hydrogen production. Renew. Sustain. Energy Rev. 2021, 145, 111064. [Google Scholar] [CrossRef]
- García-Moncada, N.; González-Castaño, M.; Ivanova, S.; Centeno, M.Á.; Romero-Sarria, F.; Odriozola, J.A. New concept for old reaction: Novel WGS catalyst design. Appl. Catal. B Environ. 2018, 238, 1–5. [Google Scholar] [CrossRef]
- Vovchok, D.; Guild, C.J.; Llorca, J.; Xu, W.; Jafari, T.; Toloueinia, P.; Kriz, D.; Waluyo, I.; Palomino, R.M.; Rodriguez, J.A.; et al. Cu supported on mesoporous ceria: Water gas shift activity at low Cu loadings through metal-support interactions. Phys. Chem. Chem. Phys. 2017, 19, 17708–17717. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.H.; Byeon, H.J.; Jang, W.J.; Jeon, K.W.; Jeong, D.W. The optimization of Nb loading amount over Cu–Nb–CeO2 catalysts for hydrogen production via the low-temperature water gas shift reaction. Int. J. Hydrogen Energy 2020, 45, 9648–9657. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Polychronopoulou, K.; Efstathiou, A.M. Novel catalytic systems for hydrogen production via the water-gas shift reaction. Conf. Pap. Energy 2013, 2013, 426980. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Gao, W. MgO-Based Intermediate-Temperature CO2 Adsorbents. In Pre-Combustion Carbon Dioxide Capture Materials; Wang, Q., Ed.; Royal Society of Chemistry: Beijing, China, 2018. [Google Scholar]
- Wang, K.; Zhao, Y.; Clough, P.T.; Zhao, P.; Anthony, E.J. Structural and kinetic analysis of CO2 sorption on NaNO2-promoted MgO at moderate temperatures. Chem. Eng. J. 2019, 372, 886–895. [Google Scholar] [CrossRef]
- Fagerlund, J.; Highfield, J.; Zevenhoven, R. Kinetics studies on wet and dry gas-solid carbonation of MgO and Mg(OH)2 for CO2 sequestration. RSC Adv. 2012, 2, 10380–10393. [Google Scholar] [CrossRef]
- Zhang, K. Development of Molten Salt Promoted Metal Oxide Based Absorbents for CO2 Separation. Ph.D. Thesis, University of Connecticut, Storrs, CT, USA, 2014. [Google Scholar]
- Dal Pozzo, A.; Armutlulu, A.; Rekhtina, M.; Abdala, P.M.; Müller, C.R. CO2 uptake and cyclic stability of MgO-based CO2 sorbents promoted with alkali metal nitrates and their eutectic mixtures. ACS Appl. Energy Mater. 2019, 2, 1295–1307. [Google Scholar] [CrossRef]
- Zhang, K.; Li, X.S.; Li, W.Z.; Rohatgi, A.; Duan, Y.; Singh, P.; Li, L.; King, D.L. Phase transfer-catalyzed fast CO2 absorption by MgO-based absorbents with high cycling capacity. Adv. Mater. Interfaces 2014, 1, 1400030. [Google Scholar] [CrossRef]
- Zhao, X.; Ji, G.; Liu, W.; He, X.; Anthony, E.J.; Zhao, M. Mesoporous MgO promoted with NaNO3/NaNO2 for rapid and high-capacity CO2 capture at moderate temperatures. Chem. Eng. J. 2018, 332, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, E.; Hassanzadeh, A.; Zarghami, S.; Arastoopour, H.; Abbasian, J. Regenerable MgO-based sorbent for high temperature CO2 removal from syngas: 3. CO2 capture and sorbent enhanced water gas shift reaction. Fuel 2014, 137, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cheng, Z.; Zhou, Z. High-purity H2 production by sorption-enhanced water gas shift on a K2CO3-promoted Cu/MgO-Al2O3 difunctional material. Sustain. Energy Fuels 2021, 5, 3340–3350. [Google Scholar] [CrossRef]
- Hiremath, V.; Trivino, M.L.T.; Seo, J.G. Eutectic mixture promoted CO2 sorption on MgO-TiO2 composite at elevated temperature. J. Environ. Sci. (China) 2019, 76, 80–88. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, L.; Hu, Y.; Cheng, Z. Nanosheet MgO-based CO2 sorbent promoted by mixed-alkali-metal nitrate and carbonate: Performance and mechanism. Ind. Eng. Chem. Res. 2017, 56, 5802–5812. [Google Scholar] [CrossRef]
- Zarghami, S.; Hassanzadeh, A.; Arastoopour, H.; Abbasian, J. Effect of steam on the reactivity of MgO-based sorbents in precombustion CO2 capture processes. Ind. Eng. Chem. Res. 2015, 54, 8860–8866. [Google Scholar] [CrossRef]
- Ryu, D.Y.; Jo, S.; Kim, T.Y.; In, S.Y.; Kim, J.K.; Hwang, J.E.; Kim, J.C.; Lee, S.C. Influence of the sorption pressure and K2CO3 loading of a MgO-based sorbent for application to the SEWGS process. Korean J. Chem. Eng. 2022, 39, 1028–1035. [Google Scholar] [CrossRef]
- Dong, H.; Yang, E.H.; Unluer, C.; Jin, F.; Al-Tabbaa, A. Investigation of the properties of MgO recovered from reject brine obtained from desalination plants. J. Clean. Prod. 2018, 196, 100–108. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Main routes for H2 production using commercial technologies: reforming, gasification and electrolysis.
Figure 1.
Main routes for H2 production using commercial technologies: reforming, gasification and electrolysis.

Figure 2.
Thermodynamic equilibrium composition as function of temperature for the WGS reaction (Reprinted with permission from [25]. 2007, Miller).
Figure 2.
Thermodynamic equilibrium composition as function of temperature for the WGS reaction (Reprinted with permission from [25]. 2007, Miller).

Figure 3.
Syngas production and main applications.

Figure 4.
Flow diagram of (a) steam methane reforming and (b) sorption-enhanced steam methane reforming (CaCO3 as sorbent).
Figure 4.
Flow diagram of (a) steam methane reforming and (b) sorption-enhanced steam methane reforming (CaCO3 as sorbent).

Figure 5.
Calcium looping diagram.

Figure 6.
Equilibrium partial pressure of CO2 from CaCO3 decomposition.

Figure 7.
Scheme of CaO and CaCO3 particles along the carbonation–calcination cycles.

Figure 8.
Scheme of (A) conventional process of the cyclic SESR, and (B) coupled process of SESR-CH4 reforming operation. (Reprinted with permission from [84]. 2021, Dang et al.).
Figure 8.
Scheme of (A) conventional process of the cyclic SESR, and (B) coupled process of SESR-CH4 reforming operation. (Reprinted with permission from [84]. 2021, Dang et al.).

Figure 9.
Scheme of the three-stage of SERP with Ca-based sorbents: pre-breakthrough stage, breakthrough stage and pre-breakthrough stage. (Reprinted with permission from [38]. 2021, Ma et al.)
Figure 9.
Scheme of the three-stage of SERP with Ca-based sorbents: pre-breakthrough stage, breakthrough stage and pre-breakthrough stage. (Reprinted with permission from [38]. 2021, Ma et al.)

Figure 10.
Carbonation–regeneration cycle of Li4SiO4 sorbent.

Figure 11.
Equilibrium partial pressure of CO2 from Li4SiO4 decomposition.

Figure 12.
Scheme of double shell structure for the CO2 sorption process in Li4SiO4. (Reprinted with permission from [113]. 2016, Yang et al.).
Figure 12.
Scheme of double shell structure for the CO2 sorption process in Li4SiO4. (Reprinted with permission from [113]. 2016, Yang et al.).

Figure 13.
Double shell model of CO2 sorption by Li2ZrO3 (a) without modification and (b) with K2CO3 addition. (Reprinted with permission from [4]. 2021, Wang et al.).
Figure 13.
Double shell model of CO2 sorption by Li2ZrO3 (a) without modification and (b) with K2CO3 addition. (Reprinted with permission from [4]. 2021, Wang et al.).

Figure 14.
Comparison of Ru/CaO, Ru/Ca3Al2O6–CaO, Ru/lime, Ca–Ni-ex-HTlc hybrid material performance: (a) CO2 capture, (b) H2 yield, (c) breakthrough curves of H2 and CH4 for Ru/CaO and Ru/Ca3Al2O6–CaO in the 1st, 5th and 10th cycles. (Reprinted with permission from [137]. 2011, Kim et al.).
Figure 14.
Comparison of Ru/CaO, Ru/Ca3Al2O6–CaO, Ru/lime, Ca–Ni-ex-HTlc hybrid material performance: (a) CO2 capture, (b) H2 yield, (c) breakthrough curves of H2 and CH4 for Ru/CaO and Ru/Ca3Al2O6–CaO in the 1st, 5th and 10th cycles. (Reprinted with permission from [137]. 2011, Kim et al.).

Figure 15.
Possible catalyst–sorbent configurations: (a) alternate layers of catalyst and sorbent, (b) alternate layers of sorbent and catalyst, (c) pre-mixed sorbent catalyst, (d) hybrid material.
Figure 15.
Possible catalyst–sorbent configurations: (a) alternate layers of catalyst and sorbent, (b) alternate layers of sorbent and catalyst, (c) pre-mixed sorbent catalyst, (d) hybrid material.

Figure 16.
Comparative scheme of WGS and SEWGS equilibrium shift. (Reprinted with permission from [167]. 2019, Gao et al.).
Figure 16.
Comparative scheme of WGS and SEWGS equilibrium shift. (Reprinted with permission from [167]. 2019, Gao et al.).

Figure 17.
Carbonation–regeneration cycle of Mg-based sorbent.

Figure 18.
MgCO3 equilibrium curve relating the temperature to the CO2 partial pressure.

Figure 19.
Effect of steam on Mg-based sorbent reactivity and capacity. (Reprinted with permission from [186]. 2015, Zarghami et al.).
Figure 19.
Effect of steam on Mg-based sorbent reactivity and capacity. (Reprinted with permission from [186]. 2015, Zarghami et al.).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
H2 production technologies, costs, efficiency, CO2 emissions and level of maturity.
| Technology | Feedstock | Production Efficiency (%) | Production Costs (€/kg H2) | CO2 Emissions (kg CO2/kg H2) | Maturity | Ref |
|---|---|---|---|---|---|---|
| SMR without CCS | Hydrocarbons | 70–85 | 0.9–2.9 | 9.2–17.2 | TRL 9 (commercial) | [2,16,17,21,23] |
| SMR with CCS | Hydrocarbons | --- | 1.7–4.1 | 2.54–9.2 | TRL 7-8 | [2,16,23] |
| Reforming with biogas | Biogas | --- | 4–6.0 | 2.93 | --- | [23] |
| Partial Oxidation | Hydrocarbons | 60–75 | --- | --- | Commercial | [17] |
| Autothermal reforming | Hydrocarbons | 60–75 | --- | --- | Near Term | [17] |
| Plasma reforming | Hydrocarbons | 8–85 | --- | --- | Long Term | [17] |
| Coal Gasification without CCS | Coal | 0.9–1.7 | 15–31 | TRL 9 (commercial) | [2,21] | |
| Coal Gasification with CCS | Coal | -- | 1.4–2.4 | 1–10 | TRL 6-7 | [2,16,21] |
| Biomass gasification | Biomass | 35–52 | 1.3–2.7 | 0.3–9 | TRL 5-6 | [2,17,21] |
| Biomass gasification with CCS | Biomass | -- | 2.8–3.2 | −11.7 to −17.5 | TRL 3-5 | [2] |
| Electrolysis | H2O + electricity | 50–70 | 4.4–8 | -- | TRL 9 Commercial | [17,23] |
| Wind Electrolysis | H2O + wind | -- | 4.01–8.8 | 0.5–1.1 | TRL 9 | [2,16,21] |
| Solar Electrolysis | H2O + sunlight | -- | 4.5–12.4 | 1.3–2.5 | TRL 9 | [2,16,21] |
| Photo Electrolysis | H2O + sunlight | 0.2 | ~ 9 | ~2 | Long Term | [16,17] |
| Thermochemical water splitting | H2O + heat | NA | -- | -- | Long Term | [17] |
Table 2.
Typical composition of syngas obtained by different processes and conditions.
| mol % (Dry Basis) | SMR [30] | Coal Gasification [31] | Indian Coal Gasification [32] | Wood Pellets Gasification [32,33] | Rice Husk Gasification [32] |
|---|---|---|---|---|---|
| H2 | 71 | 13–18 | 9 | 7–34 | 25 |
| CO2 | 6 | 7–9 | 0.6 | 6–16 | 14 |
| CO | 16 | 55–62 | 42 | 16–31 | 20 |
| O2 | -- | -- | -- | 1–3 | -- |
| N2 | -- | ~7 | 32 | 48–58 | 40 |
| CH4 | 5 | 17 | 1–4 | 0.9 | |
| CxHy | -- | -- | 0.1–0.3 | -- |
Table 3.
Main hydrocarbon reforming chemical reactions.
| Process Name | Chemical Reaction | ||
|---|---|---|---|
| Steam methane reforming | (2) | ||
| Hydrocarbon steam reforming | (3) | ||
| Organic matters steam reforming | (4) |
Table 4.
Schematic representation of the relationship between sorbent morphology and conversion without steam present, steam present during carbonation, steam during calcination and steam present for carbonation and calcination. (Reprinted with permission from [98]. 2012, Donat et al.)
Table 4.
Schematic representation of the relationship between sorbent morphology and conversion without steam present, steam present during carbonation, steam during calcination and steam present for carbonation and calcination. (Reprinted with permission from [98]. 2012, Donat et al.)
| No Steam | Steam during Calcination | Steam during Carbonation | Steam during Carbonation and Calcination |
|---|---|---|---|
 |  |  |  |
|
|
|
|
Table 5.
Carbonation reactions of alkali sorbents, theoretical CO2 uptake capacity and operating carbonation temperature range [4,29,113].
| Carbonation Reactions | ΔH⁰ 25 °C (kJ/mol) | Theoretical CO2 Uptake Capacity (g CO2/g Sorbent) | Operating Carbonation Range (°C) | |
|---|---|---|---|---|
| Li2CO3 (s) + Li2SiO3 (s) | (16) | −143 | 0.367 | 450–600 |
| Li2CO3 (s) + ZrO2 (s) | (17) | −160 | 0.288 | 450–600 |
| Na2CO3 (s) + ZrO2 (s) | (18) | −149 | 0.234 | 400–800 |
Table 6.
Properties of CaO, Li4SiO4, Li2ZrO3, KLiZrO3 and Na2ZrO3 sorbents. (Reprinted with permission from [126]. 2007, Ochoa-Fernández et al.)
Table 6.
Properties of CaO, Li4SiO4, Li2ZrO3, KLiZrO3 and Na2ZrO3 sorbents. (Reprinted with permission from [126]. 2007, Ochoa-Fernández et al.)
| CaO | Li4SiO4 | Li2ZrO3 | KliZrO3 | NaZrO3 | |
|---|---|---|---|---|---|
| Capacity | G | F | F | F | F |
| Thermodynamic | G | F | F | F | F |
| Stability | P | G | G | F | G |
| Kinetics | G | F | F/P | F | G |
G—good, F—fair, P—poor.
Table 7.
Comparison of H2 production using conventional and sorption-enhanced processes.
| Feedstock | Catalyst (CP) | Sorbent/Catalyst (SEP) | Pattern 1 | Technology 2 | Reactor 3 | Conditions | N Cycles | Maximum H2 (%) | χSE (%) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | S/C | CP | SEP | |||||||||
| CH4 | Ni/Al2O3 | Ni-CaO-Ca12Al14O33 | H | SR | FxB | 600 | 3 | 1 | ~70 | 93 | 33 | [136] |
| CH4 | Ni/Al2O3 | Ni/Al2O3-CaO | M | SR | FxB | 600 | 3 4 | ~72 | 95 98.4 | 32 37 | [148] | |
| CH4 | Ni/Al2O3 | Ni-CaO/Al2O3 Ni-CaO/Al2O3 | H H | SR | PB | 500 600 | 9 | ~60 | ~88 ~93 | 47 55 | [149] | |
| CH4 | Ni/Al2O3 Ni/Al2O3 Ni/Al2O3 | Ni/Al2O3-Li4SiO4 Ni/Al2O3-Li4SiO4 Ni/Al2O3-Li4SiO4 | M M M | SR | FB | 500 550 600 | 3.5 | 1 | 64.9 67.1 75.3 | 89.4 93.6 87.6 | 38 39 16 | [129] |
| CH4 | Ni/Al2O3 | Ni/Al2O3/K-Li4SiO4 | H | SR | FxB | 600 | 4 | 10 | ~85 | ~98 | 13 | [133] |
| CH4 | Ni commercial | Ni-Dolomite | M | SR | FB | 600 | 3 | 1 | ~73.4 | ~98 | 33 | [150] |
| Glycerol | NiO/NiAl2O4 NiO/ZrO2 | NiO/NiAl2O4 + (K-Li2ZrO3) NiO/ZrO2 + (K-Li2ZrO3) | n.a. n.a. | SR (Chemical looping) | PB | 550 | 3 | 10 | ~86 ~90 | ~90 ~93 | 5 3 | [112] |
| Ethanol | Ni/Al2O3 | Ni/Al2O3-CaO Ni/Al2O3-CaO-MgO Ni/Al2O3-Na2ZrO3 | M | SR | FxB | 600 | 6 | 1 | 64.7 | 97.0 96.2 96.5 | 50 49 49 | [11] |
| Ethanol | Ce-Ni/MCM-4 | Ce-Ni/MCM-4/Na-Zr-CaO Ce-Ni/MCM-4/Na-Zr-CaO | H M | SR | FxB | 600 | 3 | 1 | ~70 | ~94 ~80 | 34 14 | [151] |
| Ethanol | Ni/Al2O3 | Ni/Al2O3-Li4SiO 4Ni/Al2O3-K-Li4SiO4 | M M | SR | FxB | 575 | 9 | 10 | ~77 | ~98 >99 | 27 29 | [152] |
| Biogas | Ni/Al2O3 | Ni-Zr-Ca Ni-Ce-Ca Ni-La-Ca | H H H | SR | FB | 600 | 3 | 5 | 67 | ~85 ~85 ~80 | 27 27 19 | [153] |
| Bio-oil | Ce-Ni/Co-Al2O3 Ce-Ni/Co-Al2O3 Ce-Ni/Co-Al2O3 | Ce-Ni/Co-Al2O3-CaO Ce-Ni/Co-Al2O3-CaO Ce-Ni/Co-Al2O3-CaO | n.a. | SR | PB | 650 750 850 | 12 | 1 | ~65 ~70 ~75 | ~80 ~92 ~80 | 23 31 6 | [154] |
| Biogas: 50CH4/50CO2 100CH4/0CO2 | Pd/Ni-Co HT Pd/Ni-Co HT | Pd/Ni-Co HT/Dolomite Pd/Ni-Co HT/Dolomite | M | SR | FB | 600 650 | 3 | 1 | ~62 ~64 | ~98 ~98 | 58 53 | [35] |
| Biomass: corn stalk | NiO/γ-Al2O3 | NiO/γ-Al2O3-Calc. olivine NiO/γ-Al2O3-Calc. limestone NiO/γ-Al2O3-Calc. CaCO3NiO/γ-Al2O3-Calc. Dolomite | M | G | FxB | 650 | 2 | 1 | ~29 | ~47 ~50 ~67 ~72 | 62 72 131 148 | [155] |
| Coal | K2CO3 | K2CO3-Limestone | MM | G | FB | 675 | -- | 1 | ~65 | ~87 | 34 | [156] |
| Biomass: pine bark | No-catalyst CaO | No-sorbent CaO | n. a. | G | FB | 600 | -- | 1 | ~60 | ~83 | 38 | [85] |
| Biomass | Pd/Ni–Co-HT | Dolomite Pd/Ni–Co-Dolomite | M | G | FxB | 650 | -- | 1 | 69.7 | 91.1 99.0 | 31 42 | [44] |
| Biomass | Olivine | CaO-CaAl/olivine | H | G | FB | 700 | -- | 1 | 34.2 | 47.4 | 39 | [157] |
1 Mixture (M) or Hybrid (H); 2 G—gasification or SR—steam reforming; 3 FxB—Fixed Bed, PB—Packed Bed or FB—Fluidized Bed; T—temperature, S/C—steam/carbon; CP—conventional process, SEP—sorption-enhanced process; n.a.—no applicable.
Table 8.
Summary of benefits and challenges of sorption-enhanced steam reforming and gasification processes for H2 production using solid sorbents for in situ CO2 capture [4,37,113].
| Benefits | Challenges |
|---|---|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Teixeira, P.; Bacariza, C.; Correia, P.; Pinheiro, C.I.C.; Cabrita, I. Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents. Energies 2022, 15, 4039. https://doi.org/10.3390/en15114039
AMA Style
Teixeira P, Bacariza C, Correia P, Pinheiro CIC, Cabrita I. Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents. Energies. 2022; 15(11):4039. https://doi.org/10.3390/en15114039
Chicago/Turabian StyleTeixeira, Paula, Carmen Bacariza, Patrícia Correia, Carla I. C. Pinheiro, and Isabel Cabrita. 2022. "Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents" Energies 15, no. 11: 4039. https://doi.org/10.3390/en15114039
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.