

CAR-T-Derived Extracellular Vesicles: A Promising Development of CAR-T Anti-Tumor Therapy
 , , , , , , ,
, , , , , , ,  , , ,
, , ,  , , ,
, , ,  , and add
Show full author list
, and add
Show full author list
Abstract
:Simple Summary
Abstract
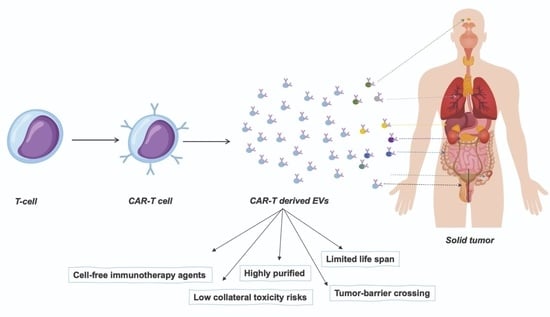
1. Introduction
2. Chimeric Antigen Receptor (CAR)-T Cell, Structure, and Evolution
3. Overview of CAR-T Cell Therapies in Hematological Malignancies
3.1. Anti CD19-Approved CAR-T Cell Therapies
3.2. Anti-BCMA Approved CAR-T Cell Therapies
4. CAR-T Cells in Solid Tumors
4.1. Ovarian Cancer
4.2. Pancreatic Cancer
4.3. Breast Cancer
4.4. Hepatocellular Carcinoma
4.5. Glioblastoma
4.6. Prostate Cancer
4.7. Renal Cancer
4.8. Gastric Cancer
4.9. Colorectal Cancer
4.10. Lung Cancer
5. Structure and Biogenesis of Extracellular Vesicles
6. T-Lymphocytes-Derived Extracellular Vesicles
7. Chimeric Antigen Receptor (CAR)-T Cell-Derived Extracellular Vesicles
8. Extracellular Vesicles as Drug Delivery Systems
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simeone, P.; Bologna, G.; Lanuti, P.; Pierdomenico, L.; Guagnano, M.T.; Pieragostino, D.; Del Boccio, P.; Vergara, D.; Marchisio, M.; Miscia, S.; et al. Extracellular Vesicles as Signaling Mediators and Disease Biomarkers across Biological Barriers. Int. J. Mol. Sci. 2020, 21, 2514. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Catitti, G.; De Bellis, D.; Vespa, S.; Simeone, P.; Canonico, B.; Lanuti, P. Extracellular Vesicles as Players in the Anti-Inflammatory Inter-Cellular Crosstalk Induced by Exercise Training. Int. J. Mol. Sci. 2022, 23, 14098. [Google Scholar] [CrossRef]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1. [Google Scholar] [CrossRef]
- Guescini, M.; Guidolin, D.; Vallorani, L.; Casadei, L.; Gioacchini, A.M.; Tibollo, P.; Battistelli, M.; Falcieri, E.; Battistin, L.; Agnati, L.F.; et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp. Cell Res. 2010, 316, 1977–1984. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Falasca, K.; Lanuti, P.; Ucciferri, C.; Pieragostino, D.; Cufaro, M.C.; Bologna, G.; Federici, L.; Miscia, S.; Pontolillo, M.; Auricchio, A.; et al. Circulating extracellular vesicles as new inflammation marker in HIV infection. AIDS 2021, 35, 595–604. [Google Scholar] [CrossRef]
- Rossi, C.; Cicalini, I.; Cufaro, M.C.; Agnifili, L.; Mastropasqua, L.; Lanuti, P.; Marchisio, M.; De Laurenzi, V.; Del Boccio, P.; Pieragostino, D. Multi-Omics Approach for Studying Tears in Treatment-Naïve Glaucoma Patients. Int. J. Mol. Sci. 2019, 20, 4029. [Google Scholar] [CrossRef]
- Brocco, D.; Lanuti, P.; Pieragostino, D.; Cufaro, M.C.; Simeone, P.; Bologna, G.; Di Marino, P.; De Tursi, M.; Grassadonia, A.; Irtelli, L.; et al. Phenotypic and Proteomic Analysis Identifies Hallmarks of Blood Circulating Extracellular Vesicles in NSCLC Responders to Immune Checkpoint Inhibitors. Cancers 2021, 13, 585. [Google Scholar] [CrossRef]
- Marchisio, M.; Simeone, P.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Pieragostino, D.; Ventrella, A.; Antonini, F.; Del Zotto, G.; Vergara, D.; et al. Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples. Int. J. Mol. Sci. 2020, 22, 48. [Google Scholar] [CrossRef]
- Pieragostino, D.; Cicalini, I.; Lanuti, P.; Ercolino, E.; di Ioia, M.; Zucchelli, M.; Zappacosta, R.; Miscia, S.; Marchisio, M.; Sacchetta, P.; et al. Enhanced release of acid sphingomyelinase-enriched exosomes generates a lipidomics signature in CSF of Multiple Sclerosis patients. Sci. Rep. 2018, 8, 3071. [Google Scholar] [CrossRef]
- Pieragostino, D.; Lanzini, M.; Cicalini, I.; Cufaro, M.C.; Damiani, V.; Mastropasqua, L.; De Laurenzi, V.; Nubile, M.; Lanuti, P.; Bologna, G.; et al. Tear proteomics reveals the molecular basis of the efficacy of human recombinant nerve growth factor treatment for Neurotrophic Keratopathy. Sci. Rep. 2022, 12, 1229. [Google Scholar] [CrossRef]
- Pieragostino, D.; Lanuti, P.; Cicalini, I.; Cufaro, M.C.; Ciccocioppo, F.; Ronci, M.; Simeone, P.; Onofrj, M.; van der Pol, E.; Fontana, A.; et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J. Proteom. 2019, 204, 103403. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Yashiro, R. Extracellular Vesicles as Novel Drug-Delivery Systems through Intracellular Communications. Membranes 2022, 12, 550. [Google Scholar] [CrossRef]
- Van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef]
- Brocco, D.; Simeone, P.; Buca, D.; Di Marino, P.; De Tursi, M.; Grassadonia, A.; De Lellis, L.; Martino, M.T.; Veschi, S.; Iezzi, M.; et al. Blood Circulating CD133+ Extracellular Vesicles Predict Clinical Outcomes in Patients with Metastatic Colorectal Cancer. Cancers 2022, 14, 1357. [Google Scholar] [CrossRef]
- Serafini, F.L.; Delli Pizzi, A.; Simeone, P.; Giammarino, A.; Mannetta, C.; Villani, M.; Izzi, J.; Buca, D.; Catitti, G.; Chiacchiaretta, P.; et al. Circulating Extracellular Vesicles: Their Role in Patients with Abdominal Aortic Aneurysm (AAA) Undergoing EndoVascular Aortic Repair (EVAR). Int. J. Mol. Sci. 2022, 23, 6015. [Google Scholar] [CrossRef] [PubMed]
- Catitti, G.; Cufaro, M.C.; De Bellis, D.; Cicalini, I.; Vespa, S.; Tonelli, F.; Miscia, G.; Secondi, L.; Simeone, P.; De Laurenzi, V.; et al. Extracellular Vesicles in Regenerative Processes Associated with Muscle Injury Recovery of Professional Athletes Undergoing Sub Maximal Strength Rehabilitation. Int. J. Mol. Sci. 2022, 23, 14913. [Google Scholar] [CrossRef] [PubMed]
- Brocco, D.; De Bellis, D.; Di Marino, P.; Simeone, P.; Grassadonia, A.; De Tursi, M.; Grottola, T.; Di Mola, F.F.; Di Gregorio, P.; Zappacosta, B.; et al. High Blood Concentration of Leukocyte-Derived Extracellular Vesicles Is Predictive of Favorable Clinical Outcomes in Patients with Pancreatic Cancer: Results from a Multicenter Prospective Study. Cancers 2022, 14, 4748. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Arzt, C.J.; Li, S.M.; Gololobova, O.A.; Batrakova, E. V Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics 2020, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef]
- Kibria, G.; Ramos, E.K.; Wan, Y.; Gius, D.R.; Liu, H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Mol. Pharm. 2018, 15, 3625–3633. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef]
- Duan, L.; Xu, L.; Xu, X.; Qin, Z.; Zhou, X.; Xiao, Y.; Liang, Y.; Xia, J. Exosome-mediated delivery of gene vectors for gene therapy. Nanoscale 2021, 13, 1387–1397. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Mirzaei, H.; Lee, S.Y.; Hadjati, J.; Till, B.G. Prospects for chimeric antigen receptor (CAR) γδ T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–423. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar] [CrossRef]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef]
- Caforio, M.; Sorino, C.; Caruana, I.; Weber, G.; Camera, A.; Cifaldi, L.; De Angelis, B.; Del Bufalo, F.; Vitale, A.; Goffredo, B.M.; et al. GD2 redirected CAR T and activated NK-cell-mediated secretion of IFNγ overcomes MYCN-dependent IDO1 inhibition, contributing to neuroblastoma cell immune escape. J. Immunother. Cancer 2021, 9, e001502. [Google Scholar] [CrossRef]
- Canzonetta, C.; Pelosi, A.; Di Matteo, S.; Veneziani, I.; Tumino, N.; Vacca, P.; Munari, E.; Pezzullo, M.; Theuer, C.; De Vito, R.; et al. Identification of neuroblastoma cell lines with uncommon TAZ+/mesenchymal stromal cell phenotype with strong suppressive activity on natural killer cells. J. Immunother. Cancer 2021, 9, e001313. [Google Scholar] [CrossRef]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Diaconu, I.; Dotti, G. From monoclonal antibodies to chimeric antigen receptors for the treatment of human malignancies. Semin. Oncol. 2014, 41, 661–666. [Google Scholar] [CrossRef]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef]
- Roselli, E.; Faramand, R.; Davila, M.L. Insight into next-generation CAR therapeutics: Designing CAR T cells to improve clinical outcomes. J. Clin. Investig. 2021, 131, e142030. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, A.; Barden, M.; Abken, H. The growing world of CAR T cell trials: A systematic review. Cancer Immunol. Immunother. 2016, 65, 1433–1450. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Buechner, J.; Caruana, I.; Künkele, A.; Rives, S.; Vettenranta, K.; Bader, P.; Peters, C.; Baruchel, A.; Calkoen, F.G. Chimeric Antigen Receptor T-Cell Therapy in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia: Curative Treatment Option or Bridge to Transplant? Front. Pediatr. 2021, 9, 784024. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-shelf” allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovich, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol. Ther. 2018, 26, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; DiPersio, J.F. Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pract. Res. Clin. Haematol. 2019, 32, 101097. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Bachmann, M.P. Conventional CARs versus modular CARs. Cancer Immunol. Immunother. 2019, 68, 1713–1719. [Google Scholar] [CrossRef]
- Martínez Bedoya, D.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022, 28, 1759–1764. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Fowler, N.H.; Dickinson, M.; Dreyling, M.; Martinez-Lopez, J.; Kolstad, A.; Butler, J.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E.; et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: The phase 2 ELARA trial. Nat. Med. 2022, 28, 325–332. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Ogasawara, K.; Dodds, M.; Mack, T.; Lymp, J.; Dell’Aringa, J.; Smith, J. Population Cellular Kinetics of Lisocabtagene Maraleucel, an Autologous CD19-Directed Chimeric Antigen Receptor T-Cell Product, in Patients with Relapsed/Refractory Large B-Cell Lymphoma. Clin. Pharmacokinet. 2021, 60, 1621–1633. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2022, JCO.22.00842. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef]
- Pellegrino, M.; Del Bufalo, F.; De Angelis, B.; Quintarelli, C.; Caruana, I.; de Billy, E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells 2020, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Simula, L.; Locatelli, F.; Campello, S. T lymphocytes against solid malignancies: Winning ways to defeat tumours. Cell Stress 2018, 2, 200–212. [Google Scholar] [CrossRef]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef]
- Min, I.M.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Wyrwas, B.; Scognamiglio, T.; Moore, M.D.; Wang, W.; Park, S.; Park, S.; et al. CAR T Therapy Targeting ICAM-1 Eliminates Advanced Human Thyroid Tumors. Clin. Cancer Res. 2017, 23, 7569–7583. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.-M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef]
- Magee, M.S.; Abraham, T.S.; Baybutt, T.R.; Flickinger, J.C.; Ridge, N.A.; Marszalowicz, G.P.; Prajapati, P.; Hersperger, A.R.; Waldman, S.A.; Snook, A.E. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol. Res. 2018, 6, 509–516. [Google Scholar] [CrossRef]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.-C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72+ Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef]
- Chekmasova, A.A.; Rao, T.D.; Nikhamin, Y.; Park, K.J.; Levine, D.A.; Spriggs, D.R.; Brentjens, R.J. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 2010, 16, 3594–3606. [Google Scholar] [CrossRef]
- Owens, G.L.; Sheard, V.E.; Kalaitsidou, M.; Blount, D.; Lad, Y.; Cheadle, E.J.; Edmondson, R.J.; Kooner, G.; Gilham, D.E.; Harrop, R. Preclinical Assessment of CAR T-Cell Therapy Targeting the Tumor Antigen 5T4 in Ovarian Cancer. J. Immunother. 2018, 41, 130–140. [Google Scholar] [CrossRef]
- Zuo, S.; Wen, Y.; Panha, H.; Dai, G.; Wang, L.; Ren, X.; Fu, K. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol. Immunol. 2017, 85, 293–304. [Google Scholar] [CrossRef]
- Schäfer, D.; Tomiuk, S.; Küster, L.N.; Al Rawashdeh, W.; Henze, J.; Tischler-Höhle, G.; Agorku, D.J.; Brauner, J.; Linnartz, C.; Lock, D.; et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat. Commun. 2021, 12, 1453. [Google Scholar] [CrossRef]
- Cutmore, L.C.; Brown, N.F.; Raj, D.; Chauduri, S.; Wang, P.; Maher, J.; Wang, Y.; Lemoine, N.R.; Marshall, J.F. Pancreatic Cancer UK Grand Challenge: Developments and challenges for effective CAR T cell therapy for pancreatic ductal adenocarcinoma. Pancreatology 2020, 20, 394–408. [Google Scholar] [CrossRef]
- Tomar, S.; Zhang, J.; Khanal, M.; Hong, J.; Venugopalan, A.; Jiang, Q.; Sengupta, M.; Miettinen, M.; Li, N.; Pastan, I.; et al. Development of Highly Effective Anti-Mesothelin hYP218 Chimeric Antigen Receptor T Cells With Increased Tumor Infiltration and Persistence for Treating Solid Tumors. Mol. Cancer Ther. 2022, 21, 1195–1206. [Google Scholar] [CrossRef]
- Raj, D.; Nikolaidi, M.; Garces, I.; Lorizio, D.; Castro, N.M.; Caiafa, S.G.; Moore, K.; Brown, N.F.; Kocher, H.M.; Duan, X.; et al. CEACAM7 Is an Effective Target for CAR T-cell Therapy of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1538–1552. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Zhou, Y.; Wen, P.; Li, M.; Li, Y.; Li, X.-A. Construction of chimeric antigen receptor-modified T cells targeting EpCAM and assessment of their anti-tumor effect on cancer cells. Mol. Med. Rep. 2019, 20, 2355–2364. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, Q. Recent updates on chimeric antigen receptor T cell therapy for hepatocellular carcinoma. Cancer Gene Ther. 2021, 28, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yang, D.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef]
- Soler, D.C.; Kerstetter-Fogle, A.; McCormick, T.S.; Sloan, A.E. Using chimeric antigen receptor T-cell therapy to fight glioblastoma multiforme: Past, present and future developments. J. Neurooncol. 2022, 156, 81–96. [Google Scholar] [CrossRef]
- Chow, K.K.H.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.-F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef]
- An, Z.; Hu, Y.; Bai, Y.; Zhang, C.; Xu, C.; Kang, X.; Yang, S.; Li, W.; Zhong, X. Antitumor activity of the third generation EphA2 CAR-T cells against glioblastoma is associated with interferon gamma induced PD-L1. Oncoimmunology 2021, 10, 1960728. [Google Scholar] [CrossRef]
- De Billy, E.; Pellegrino, M.; Orlando, D.; Pericoli, G.; Ferretti, R.; Businaro, P.; Ajmone-Cat, M.A.; Rossi, S.; Petrilli, L.L.; Maestro, N.; et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro. Oncol. 2022, 24, 1150–1163. [Google Scholar] [CrossRef]
- Arnone, C.M.; Polito, V.A.; Mastronuzzi, A.; Carai, A.; Diomedi, F.C.; Antonucci, L.; Petrilli, L.L.; Vinci, M.; Ferrari, F.; Salviato, E.; et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J. Immunother. Cancer 2021, 9, e001930. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, Q.; Zhang, J.; Liu, F. Targeting Tumor-Associated Antigen: A Promising CAR-T Therapeutic Strategy for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 661606. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maker, M.; Aftabizadeh, M.; Yang, X.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ Is Critical for CAR T Cell-Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021, 11, 2248–2265. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef]
- Weimin, S.; Abula, A.; Qianghong, D.; Wenguang, W. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol. Ther. 2020, 21, 570–580. [Google Scholar] [CrossRef]
- Jiang, Y.; Wen, W.; Yang, F.; Han, D.; Zhang, W.; Qin, W. Prospect of Prostate Cancer Treatment: Armed CAR-T or Combination Therapy. Cancers 2022, 14, 967. [Google Scholar] [CrossRef]
- Lloyd, G.G.; Steel, J.M.; Young, R.J. Eating disorders and psychiatric morbidity in patients with diabetes mellitus. Psychother. Psychosom. 1987, 48, 189–195. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.Y.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Bagley, S.J.; O’Rourke, D.M. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Bui, M.H.T.; Seligson, D.; Han, K.; Pantuck, A.J.; Dorey, F.J.; Huang, Y.; Horvath, S.; Leibovich, B.C.; Chopra, S.; Liao, S.-Y.; et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: Implications for prognosis and therapy. Clin. Cancer Res. 2003, 9, 802–811. [Google Scholar]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-Cell Therapy for Solid Tumors. Am. Soc. Clin. Oncol. Educ. book. Am. Soc. Clin. Oncol. Annu. Meet. 2017, 37, 193–204. [Google Scholar] [CrossRef]
- Tafreshi, N.K.; Lloyd, M.C.; Bui, M.M.; Gillies, R.J.; Morse, D.L. Carbonic anhydrase IX as an imaging and therapeutic target for tumors and metastases. Subcell. Biochem. 2014, 75, 221–254. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Jia, L.; Zhang, M.; Huang, X.; Qian, P.; Tang, Q.; Zhu, J.; Feng, Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am. J. Cancer Res. 2019, 9, 1846–1856. [Google Scholar] [PubMed]
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shi, Z.; Wang, P.; Wang, C.; Yang, L.; Du, G.; Zhang, H.; Shi, B.; Jia, J.; Li, Q.; et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019, 111, 409–418. [Google Scholar] [CrossRef]
- Tao, K.; He, M.; Tao, F.; Xu, G.; Ye, M.; Zheng, Y.; Li, Y. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother. Pharmacol. 2018, 82, 815–827. [Google Scholar] [CrossRef]
- Kim, M.; Pyo, S.; Kang, C.H.; Lee, C.O.; Lee, H.K.; Choi, S.U.; Park, C.H. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS ONE 2018, 13, e0198347. [Google Scholar] [CrossRef]
- Song, Y.; Tong, C.; Wang, Y.; Gao, Y.; Dai, H.; Guo, Y.; Zhao, X.; Wang, Y.; Wang, Z.; Han, W.; et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell 2018, 9, 867–878. [Google Scholar] [CrossRef]
- Jung, M.; Yang, Y.; McCloskey, J.E.; Zaman, M.; Vedvyas, Y.; Zhang, X.; Stefanova, D.; Gray, K.D.; Min, I.M.; Zarnegar, R.; et al. Chimeric Antigen Receptor T Cell Therapy Targeting ICAM-1 in Gastric Cancer. Mol. Ther. Oncolytics 2020, 18, 587–601. [Google Scholar] [CrossRef]
- Deng, X.; Gao, F.; Li, N.; Li, Q.; Zhou, Y.; Yang, T.; Cai, Z.; Du, P.; Chen, F.; Cai, J. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am. J. Cancer Res. 2019, 9, 945–958. [Google Scholar]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-Menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2019, 12, 54. [Google Scholar] [CrossRef]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, J.; Wu, X.; Zhang, M.; Luo, D.; Zhang, R.; Li, S.; He, Y.; Bian, H.; Chen, Z. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Front. Med. 2019, 13, 57–68. [Google Scholar] [CrossRef]
- Ye, L.; Lou, Y.; Lu, L.; Fan, X. Mesothelin-targeted second generation CAR-T cells inhibit growth of mesothelin-expressing tumors in vivo. Exp. Ther. Med. 2019, 17, 739–747. [Google Scholar] [CrossRef]
- Li, N.; Liu, S.; Sun, M.; Chen, W.; Xu, X.; Zeng, Z.; Tang, Y.; Dong, Y.; Chang, A.H.; Zhao, Q. Chimeric Antigen Receptor-Modified T Cells Redirected to EphA2 for the Immunotherapy of Non-Small Cell Lung Cancer. Transl. Oncol. 2018, 11, 11–17. [Google Scholar] [CrossRef]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Li, W.; Yu, X.; Flores-Villanueva, P.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; Young, K.H.; Ma, X.; et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 2020, 9, 72. [Google Scholar] [CrossRef]
- Chen, X.; Amar, N.; Zhu, Y.; Wang, C.; Xia, C.; Yang, X.; Wu, D.; Feng, M. Combined DLL3-targeted bispecific antibody with PD-1 inhibition is efficient to suppress small cell lung cancer growth. J. Immunother. Cancer 2020, 8, e000785. [Google Scholar] [CrossRef]
- Aharon, A.; Horn, G.; Bar-Lev, T.H.; Zagagi Yohay, E.; Waks, T.; Levin, M.; Deshet Unger, N.; Avivi, I.; Globerson Levin, A. Extracellular Vesicles Derived from Chimeric Antigen Receptor-T Cells: A Potential Therapy for Cancer. Hum. Gene Ther. 2021, 32, 1224–1241. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell. Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.L.; Rood, I.M.; Deegens, J.K.J.; Klein, J.B. Isolation and characterization of urinary extracellular vesicles: Implications for biomarker discovery. Nat. Rev. Nephrol. 2017, 13, 731–749. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Graves-Deal, R.; Trinh, V.Q.; Ramirez, M.A.; Sohn, Y.; Neininger, A.C.; Taneja, N.; McKinley, E.T.; et al. Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nat. Cell Biol. 2021, 23, 1240–1254. [Google Scholar] [CrossRef]
- Yu, S.; Yu, L. Migrasome biogenesis and functions. FEBS J. 2022, 289, 7246–7254. [Google Scholar] [CrossRef]
- De Lellis, L.; Florio, R.; Di Bella, M.C.; Brocco, D.; Guidotti, F.; Tinari, N.; Grassadonia, A.; Lattanzio, R.; Cama, A.; Veschi, S. Exosomes as Pleiotropic Players in Pancreatic Cancer. Biomedicines 2021, 9, 275. [Google Scholar] [CrossRef]
- Clancy, J.W.; Boomgarden, A.C.; D’Souza-Schorey, C. Profiling and promise of supermeres. Nat. Cell Biol. 2021, 23, 1217–1219. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef]
- Tamai, K.; Tanaka, N.; Nakano, T.; Kakazu, E.; Kondo, Y.; Inoue, J.; Shiina, M.; Fukushima, K.; Hoshino, T.; Sano, K.; et al. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem. Biophys. Res. Commun. 2010, 399, 384–390. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Buschow, S.I.; Nolte-’t Hoen, E.N.M.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.M.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Venn, G.; Lauder, R.M.; Hardingham, T.E.; Muir, H. Effects of catabolic and anabolic cytokines on proteoglycan biosynthesis in young, old and osteoarthritic canine cartilage. Biochem. Soc. Trans. 1990, 18, 973–974. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Bergam, P.; Di Cicco, A.; Hurbain, I.; Lo Cicero, A.; Dingli, F.; Palmulli, R.; Fort, C.; Potier, M.C.; Schurgers, L.J.; et al. Apolipoprotein E Regulates Amyloid Formation within Endosomes of Pigment Cells. Cell Rep. 2015, 13, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.A.; Pérez-González, R.; Sharma, A.; Huang, F.-K.; Alldred, M.J.; Pawlik, M.; Kaur, G.; Ginsberg, S.D.; Neubert, T.A.; Levy, E. Enhanced exosome secretion in Down syndrome brain—A protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Calvo, V.; Izquierdo, M. Imaging Polarized Secretory Traffic at the Immune Synapse in Living T Lymphocytes. Front. Immunol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Mathivanan, S.; Ji, H.; Simpson, R.J. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol. Cell. Proteomics 2013, 12, 587–598. [Google Scholar] [CrossRef]
- Pallet, N.; Sirois, I.; Bell, C.; Hanafi, L.-A.; Hamelin, K.; Dieudé, M.; Rondeau, C.; Thibault, P.; Desjardins, M.; Hebert, M.-J. A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics 2013, 13, 1108–1120. [Google Scholar] [CrossRef]
- Brocco, D.; Lanuti, P.; Simeone, P.; Bologna, G.; Pieragostino, D.; Cufaro, M.C.; Graziano, V.; Peri, M.; Di Marino, P.; De Tursi, M.; et al. Circulating Cancer Stem Cell-Derived Extracellular Vesicles as a Novel Biomarker for Clinical Outcome Evaluation. J. Oncol. 2019, 2019, 5879616. [Google Scholar] [CrossRef]
- Buca, D.; D’Antonio, F.; Buca, D.; Di Sebastiano, F.; Simeone, P.; Di Girolamo, R.; Bologna, G.; Vespa, S.; Catitti, G.; Liberati, M.; et al. Extracellular Vesicles in pregnancy: Their potential role as a liquid biopsy. J. Reprod. Immunol. 2022, 154, 103734. [Google Scholar] [CrossRef]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; Ní Áinle, F.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles From Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk With Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef]
- Lanuti, P.; Santilli, F.; Marchisio, M.; Pierdomenico, L.; Vitacolonna, E.; Santavenere, E.; Iacone, A.; Davì, G.; Romano, M.; Miscia, S. A novel flow cytometric approach to distinguish circulating endothelial cells from endothelial microparticles: Relevance for the evaluation of endothelial dysfunction. J. Immunol. Methods 2012, 380, 16–22. [Google Scholar] [CrossRef]
- Simeone, P.; Celia, C.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Cilurzo, F.; Grande, R.; Diomede, F.; Vespa, S.; Canonico, B.; et al. Diameters and Fluorescence Calibration for Extracellular Vesicle Analyses by Flow Cytometry. Int. J. Mol. Sci. 2020, 21, 7885. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Soon, R.H.; Zhang, P.; Jiang, K.; Lim, C.T. Cancer diagnosis: From tumor to liquid biopsy and beyond. Lab Chip 2018, 19, 11–34. [Google Scholar] [CrossRef]
- Buca, D.; Bologna, G.; D’Amico, A.; Cugini, S.; Musca, F.; Febbo, M.; D’Arcangelo, D.; Buca, D.; Simeone, P.; Liberati, M.; et al. Extracellular Vesicles in Feto-Maternal Crosstalk and Pregnancy Disorders. Int. J. Mol. Sci. 2020, 21, 2120. [Google Scholar] [CrossRef]
- Serafini, F.L.; Lanuti, P.; Delli Pizzi, A.; Procaccini, L.; Villani, M.; Taraschi, A.L.; Pascucci, L.; Mincuzzi, E.; Izzi, J.; Chiacchiaretta, P.; et al. Diagnostic Impact of Radiological Findings and Extracellular Vesicles: Are We Close to Radiovesicolomics? Biology 2021, 10, 1265. [Google Scholar] [CrossRef]
- Lucidi, A.; Buca, D.; Ronsini, C.; Tinari, S.; Bologna, G.; Buca, D.; Leombroni, M.; Liberati, M.; D’Antonio, F.; Scambia, G.; et al. Role of Extracellular Vesicles in Epithelial Ovarian Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 8762. [Google Scholar] [CrossRef]
- Mack, M.; Kleinschmidt, A.; Brühl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachý, J.; Stangassinger, M.; Erfle, V.; Schlöndorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.-P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.-K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Tanaka, M.; Itai, T.; Adachi, M.; Nagata, S. Downregulation of Fas ligand by shedding. Nat. Med. 1998, 4, 31–36. [Google Scholar] [CrossRef]
- Schneider, P.; Holler, N.; Bodmer, J.L.; Hahne, M.; Frei, K.; Fontana, A.; Tschopp, J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 1998, 187, 1205–1213. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Oh, N.; Park, J.-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomedicine 2014, 9 (Suppl. S1), 51–63. [Google Scholar] [CrossRef]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J. Extracell. Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Antes, T.J.; Middleton, R.C.; Luther, K.M.; Ijichi, T.; Peck, K.A.; Liu, W.J.; Valle, J.; Echavez, A.K.; Marbán, E. Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. J. Nanobiotechnology 2018, 16, 61. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Meckes, D.G. Extracellular Vesicle Integrins Distinguish Unique Cancers. Proteomes 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell. Biochem. 2010, 111, 488–496. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Hazrati, A.; Soudi, S.; Malekpour, K.; Mahmoudi, M.; Rahimi, A.; Hashemi, S.M.; Varma, R.S. Immune cells-derived exosomes function as a double-edged sword: Role in disease progression and their therapeutic applications. Biomark. Res. 2022, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Seebauer, C.; Wende, K.; Schmidt, A. Physical plasma and leukocytes—Immune or reactive? Biol. Chem. 2018, 400, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Pugholm, L.H.; Bæk, R.; Søndergaard, E.K.L.; Revenfeld, A.L.S.; Jørgensen, M.M.; Varming, K. Phenotyping of Leukocytes and Leukocyte-Derived Extracellular Vesicles. J. Immunol. Res. 2016, 2016, 6391264. [Google Scholar] [CrossRef]
- Calvo, V.; Izquierdo, M. Inducible Polarized Secretion of Exosomes in T and B Lymphocytes. Int. J. Mol. Sci. 2020, 21, 2631. [Google Scholar] [CrossRef]
- Van der Vlist, E.J.; Arkesteijn, G.J.A.; van de Lest, C.H.A.; Stoorvogel, W.; Nolte-’t Hoen, E.N.M.; Wauben, M.H.M. CD4(+) T cell activation promotes the differential release of distinct populations of nanosized vesicles. J. Extracell. Vesicles 2012, 1, 18364. [Google Scholar] [CrossRef]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and Function of T Cell-Derived Exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Herz, J.; Pardo, J.; Kashkar, H.; Schramm, M.; Kuzmenkina, E.; Bos, E.; Wiegmann, K.; Wallich, R.; Peters, P.J.; Herzig, S.; et al. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat. Immunol. 2009, 10, 761–768. [Google Scholar] [CrossRef]
- Alonso, R.; Mazzeo, C.; Mérida, I.; Izquierdo, M. A new role of diacylglycerol kinase alpha on the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. Biochimie 2007, 89, 213–221. [Google Scholar] [CrossRef]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Rodríguez, M.C.; Pindado, J.; Merino, E.; Mérida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. J. Biol. Chem. 2005, 280, 28439–28450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, M.A.; Jones, D.R.; Izquierdo, M.; Mérida, I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, X.; Zhao, T.; Li, W.; Xiang, J. Natural CD8+25+ regulatory T cell-secreted exosomes capable of suppressing cytotoxic T lymphocyte-mediated immunity against B16 melanoma. Biochem. Biophys. Res. Commun. 2013, 438, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef]
- Martínez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monlen, I.; Lasierra, P.; Larrad, L.; Piñeiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar] [CrossRef]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef]
- Choudhuri, K.; Llodrá, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef]
- Saliba, D.G.; Céspedes-Donoso, P.F.; Bálint, Š.; Compeer, E.B.; Korobchevskaya, K.; Valvo, S.; Mayya, V.; Kvalvaag, A.; Peng, Y.; Dong, T.; et al. Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. Elife 2019, 8, e47528. [Google Scholar] [CrossRef]
- Peters, P.J.; Geuze, H.J.; Van der Donk, H.A.; Slot, J.W.; Griffith, J.M.; Stam, N.J.; Clevers, H.C.; Borst, J. Molecules relevant for T cell-target cell interaction are present in cytolytic granules of human T lymphocytes. Eur. J. Immunol. 1989, 19, 1469–1475. [Google Scholar] [CrossRef]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krähenbühl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef]
- Tang, X.-J.; Sun, X.-Y.; Huang, K.-M.; Zhang, L.; Yang, Z.-S.; Zou, D.-D.; Wang, B.; Warnock, G.L.; Dai, L.-J.; Luo, J. Therapeutic potential of CAR-T cell-derived exosomes: A cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef]
- Johnson, L.R.; Lee, D.Y.; Eacret, J.S.; Ye, D.; June, C.H.; Minn, A.J. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell 2021, 184, 4981–4995.e14. [Google Scholar] [CrossRef]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef]
- Del Vecchio, F.; Martinez-Rodriguez, V.; Schukking, M.; Cocks, A.; Broseghini, E.; Fabbri, M. Professional killers: The role of extracellular vesicles in the reciprocal interactions between natural killer, CD8+ cytotoxic T-cells and tumour cells. J. Extracell. Vesicles 2021, 10, e12075. [Google Scholar] [CrossRef]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef]
- Wu, C.-H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef]
- Wu, F.; Xie, M.; Hun, M.; She, Z.; Li, C.; Luo, S.; Chen, X.; Wan, W.; Wen, C.; Tian, J. Natural Killer Cell-Derived Extracellular Vesicles: Novel Players in Cancer Immunotherapy. Front. Immunol. 2021, 12, 658698. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Marschall, A.L.J. Targeting the Inside of Cells with Biologicals: Chemicals as a Delivery Strategy. BioDrugs 2021, 35, 643–671. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Wolfram, J. Extracellular vesicles versus synthetic nanoparticles for drug delivery. Nat. Rev. Mater. 2021, 6, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Gangadaran, P.; Li, X.J.; Lee, H.W.; Oh, J.M.; Kalimuthu, S.; Rajendran, R.L.; Son, S.H.; Baek, S.H.; Singh, T.D.; Zhu, L.; et al. A new bioluminescent reporter system to study the biodistribution of systematically injected tumor-derived bioluminescent extracellular vesicles in mice. Oncotarget 2017, 8, 109894–109914. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Reshke, R.; Taylor, J.A.; Savard, A.; Guo, H.; Rhym, L.H.; Kowalski, P.S.; Trung, M.T.; Campbell, C.; Little, W.; Anderson, D.G.; et al. Reduction of the therapeutic dose of silencing RNA by packaging it in extracellular vesicles via a pre-microRNA backbone. Nat. Biomed. Eng. 2020, 4, 52–68. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Malekian, F.; Shamsian, A.; Kodam, S.P.; Ullah, M. Exosome engineering for efficient and targeted drug delivery: Current status and future perspective. J. Physiol. 2022, in press. [Google Scholar] [CrossRef]
- Rankin-Turner, S.; Vader, P.; O’Driscoll, L.; Giebel, B.; Heaney, L.M.; Davies, O.G. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Adv. Drug Deliv. Rev. 2021, 173, 479–491. [Google Scholar] [CrossRef]
- Chen, C.; Sun, M.; Wang, J.; Su, L.; Lin, J.; Yan, X. Active cargo loading into extracellular vesicles: Highlights the heterogeneous encapsulation behaviour. J. Extracell. Vesicles 2021, 10, e12163. [Google Scholar] [CrossRef]
- Görgens, A.; Corso, G.; Hagey, D.W.; Jawad Wiklander, R.; Gustafsson, M.O.; Felldin, U.; Lee, Y.; Bostancioglu, R.B.; Sork, H.; Liang, X.; et al. Identification of storage conditions stabilizing extracellular vesicles preparations. J. Extracell. Vesicles 2022, 11, e12238. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Targets Used for CAR-T Cell Production |
|---|---|
| Ovarian Cancer | TAG72 [90]; MUC16 [91]; HER2 * [91] [141]; 5T4 [92]; FRα [93] |
| Pancreatic Cancer | MSLN [95,96]; CEACAM7 [97]; CEACAM6, CD318, TSPAN8 [94] |
| Breast Cancer | MSLN * [98,99,142]; EpCAM [100]; HER2 * [141,143]; |
| Hepatocellular Carcinoma | GPC3 [101]; AFP [102]; NKG2DL [103] |
| Glioblastoma | IL13Rα2 [110,111]; HER2 [112]; EphA2 [106,107]; EGFRvIII [105] |
| Prostate Cancer | PSMA [115,116]; PSCA [114]; EpCAM [114] |
| Renal Cancer | CA-IX [121] |
| Gastric Cancer | Trop2/PD-L1 [122]; MSLN [123]; FOLR1 [126]; claudin 18.2 [124]; HER2 [127]; NKG2D [125]; ICAM-1 [128] |
| Colorectal Cancer | CD133 [130]; TAG-72 [131]; NKG2D [129]; Guanylate Cyclase 2C [89]; CEA [132]; DCLK1 [133] |
| Lung Cancer | ROR1 [134]; EGFRvIII [135]; EphA2 [137]; MSLN [136]; mucin-1 [138]; PSCA [138]; PD-L1 [139]; DLL3 [140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagotto, S.; Simeone, P.; Brocco, D.; Catitti, G.; De Bellis, D.; Vespa, S.; Di Pietro, N.; Marinelli, L.; Di Stefano, A.; Veschi, S.; et al. CAR-T-Derived Extracellular Vesicles: A Promising Development of CAR-T Anti-Tumor Therapy. Cancers 2023, 15, 1052. https://doi.org/10.3390/cancers15041052
Pagotto S, Simeone P, Brocco D, Catitti G, De Bellis D, Vespa S, Di Pietro N, Marinelli L, Di Stefano A, Veschi S, et al. CAR-T-Derived Extracellular Vesicles: A Promising Development of CAR-T Anti-Tumor Therapy. Cancers. 2023; 15(4):1052. https://doi.org/10.3390/cancers15041052
Chicago/Turabian StylePagotto, Sara, Pasquale Simeone, Davide Brocco, Giulia Catitti, Domenico De Bellis, Simone Vespa, Natalia Di Pietro, Lisa Marinelli, Antonio Di Stefano, Serena Veschi, and et al. 2023. "CAR-T-Derived Extracellular Vesicles: A Promising Development of CAR-T Anti-Tumor Therapy" Cancers 15, no. 4: 1052. https://doi.org/10.3390/cancers15041052