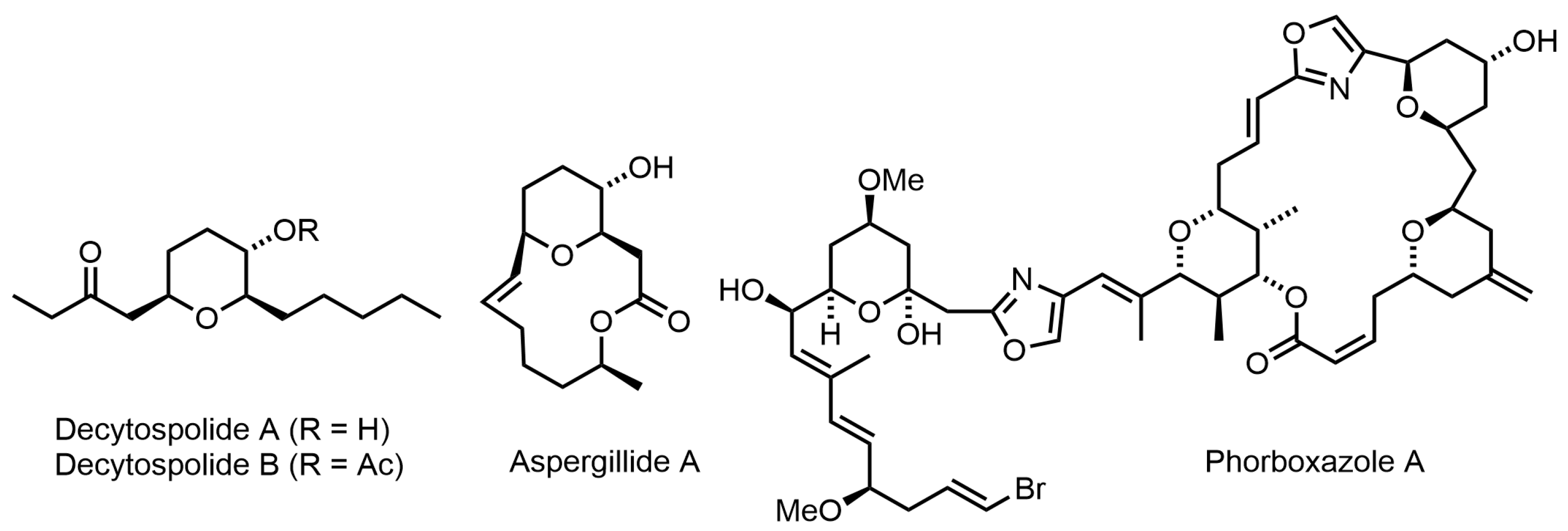
2. Results and Discussion
We began by investigating the gold-catalyzed sequential reaction with bis-propargylic alcohol
13a as a model substrate in the presence of various gold (I) catalysts (
Table 1).
![Catalysts 14 00228 i001]()
Treatment of bis-propargyl alcohol
13a with MeOH (2 eq.) in the presence of Ph
3PAuNTf
2 (10 mol%) in toluene at reflux afforded a moderate yield of the product
14′a (
cis:
trans = 1:1) without the formation of the desired 2,6-
cis-disubstituted tetrahydropyran
14a (entry 1). The reaction with the activated gold (I) species generated from Ph
3PAuCl (10 mol%) or (C
6F
5)
3PAuCl (10 mol%) by the silver catalyst AgNTf
2 (10 mol%) furnished the product
14′a in 74% yield (
cis:
trans = 1:1) and 68% yield (
cis:
trans = 2:1), respectively, without the desired product
14a (entries 2 and 3). The reaction with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in ClCH
2CH
2Cl as solvent instead of toluene furnished the desired product
14a in 37% yield, along with 14% yield of the product
14′a (entry 4). The reaction in ClCH
2CH
2Cl with H
2O (10 eq.) to accelerate the oxa-Michael addition reaction afforded the desired
cis-2,6-disubstituted tetrahydropyran
14a in 56% yield (entry 5), while the reaction with H
2O (10 eq.) in toluene furnished 28% yield of the tetrahydropyran
14′a without the desired
cis-2,6-disubtituted tetrahydropyran
14a (entry 6). Changing the reaction solvent from toluene to 1,2-dichloroethane gave the desired
cis-2,6-disubstituted tetrahydropyran
14a, probably due to the difference in water solubility of the solvents. Thus, the solubility of water (8.60 g/L, 25 °C) [
25] in 1,2-dichloroethane is higher than that of toluene (515 mg/L, 20 °C) [
26], which suggests that water is involved in the reaction in the dichloroethane to obtain the desired product. On the other hand, the reaction with AuBr
3 (10 mol%) in 1,2-dichloroethane at reflux for 48 h gave 2,6-disubstituted tetrahydropyran
14″a in 46% yield (
cis:
trans = 1:1) (entry 7). Finally, the optimal reaction conditions for preparation of the desired product
14a from bis-propargylic alcohol
13a were found to be Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of MeOH (2 eq.) and H
2O (10 eq.) in ClCH
2CH
2Cl stirred at 50 °C.
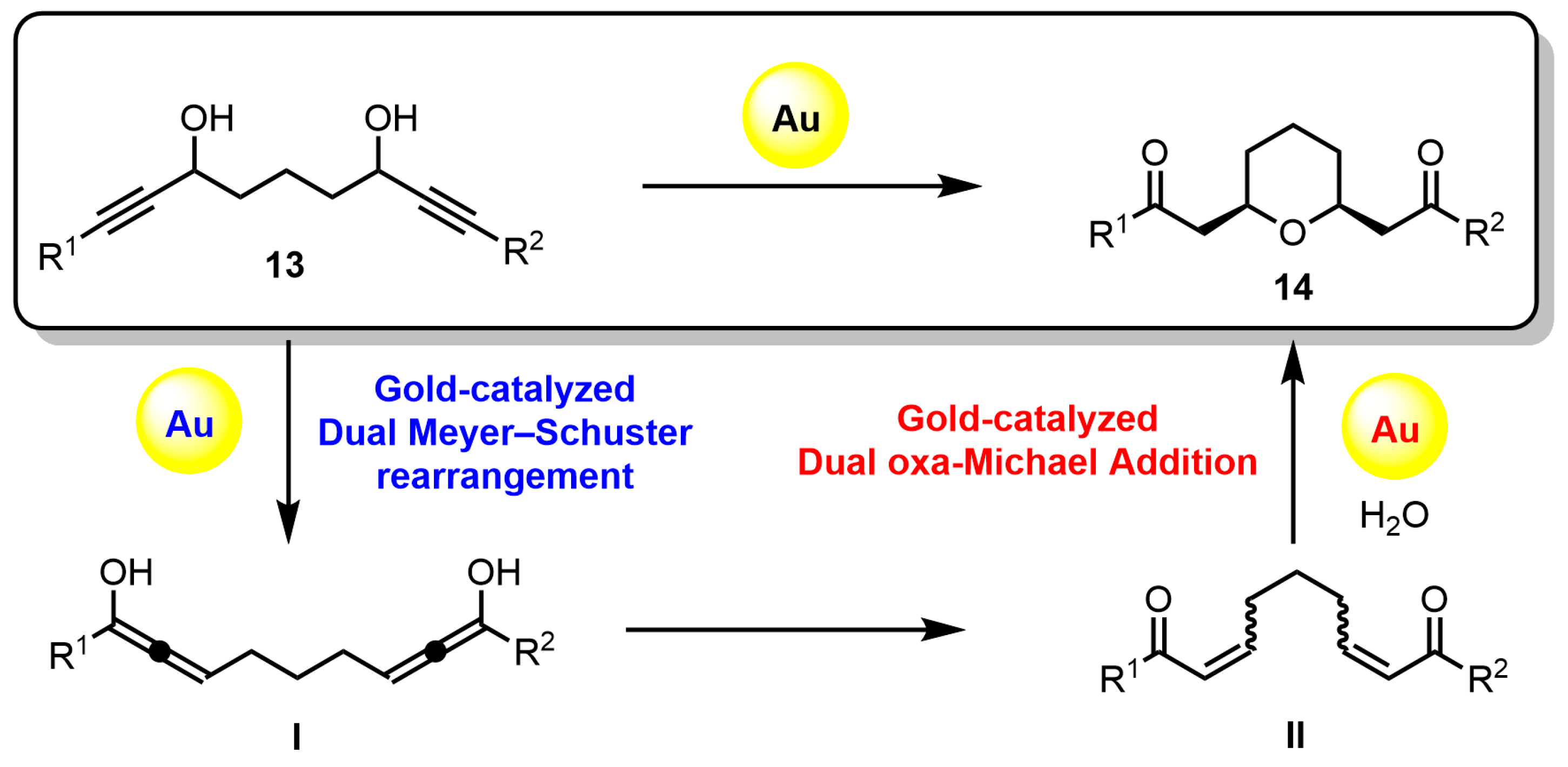
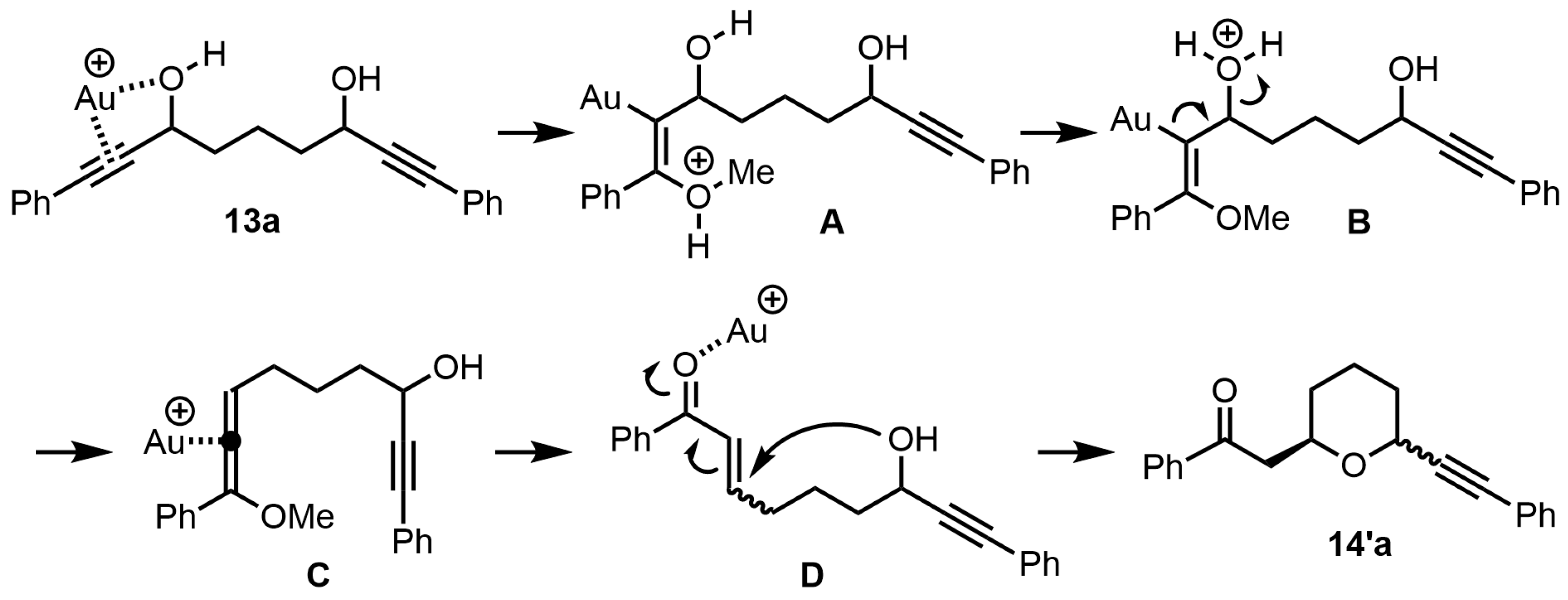
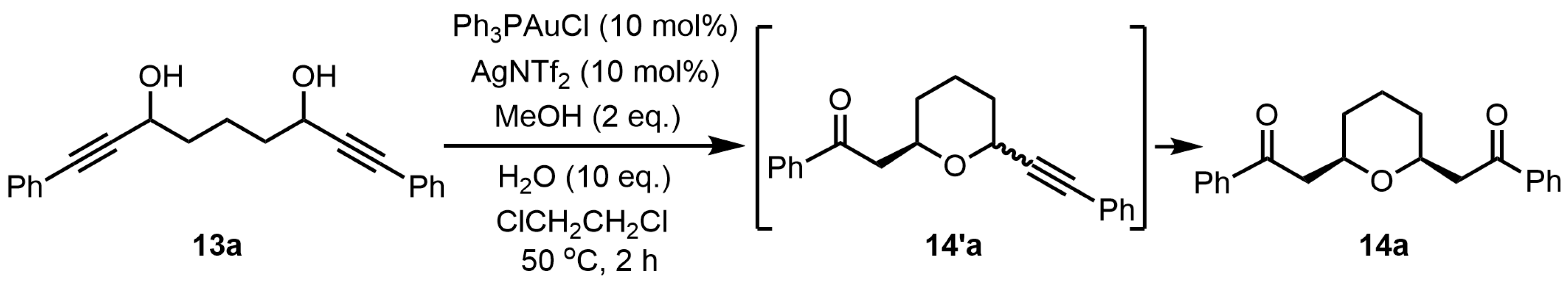
From this result, the plausible reaction mechanism for the preparation of 2,6-disubstituted tetrahydropyran
14′a was shown in
Scheme 3. It is assumed that the tetrahydropyran
14′a is formed by the Meyer–Schuster rearrangement reaction followed by the intramolecular oxa-Michael addition reaction. First, the gold (I) catalyst is coordinated to the triple bond and hydroxyl group on one side of bis-propargylic alcohol
13a, resulting in the addition of methanol to the activated triple bond by gold (I) to afford the allenyl ether
C (
13a→
A→
B→
C). Then, hydrolysis of the allenyl ether
C gives α,β-unsaturated ketone
D, which undergoes oxa-Michael addition to furnish the tetrahydropyran
14′a (
C→
D→
14′a). However, it was not clear whether the mechanism of formation for the desired
cis-2,6-disubstituted tetrahydropyran
14a was from the tetrahydropyran
14′a or some other mechanism.
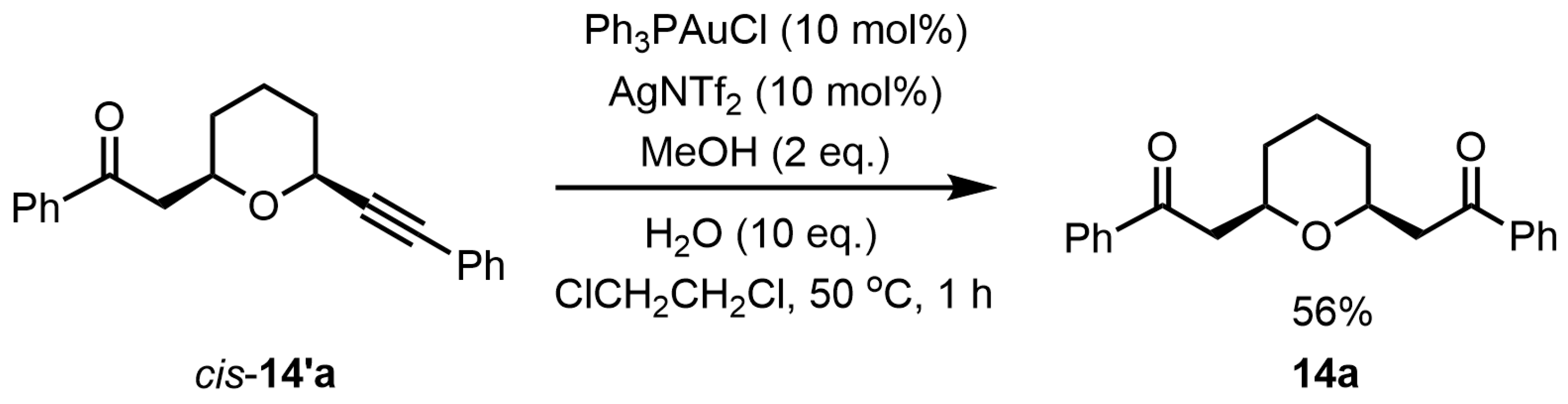
Next, to elucidate the mechanism of formation of the desired
cis-2,6-disubstituted tetrahydropyran
14a, the reaction of
cis-disubstituted tetrahydropyran
14′a was performed under optimal reaction conditions (
Scheme 4). Treatment of tetrahydropyran
14′a with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of MeOH (2 eq.) and H
2O (10 eq.) in ClCH
2CH
2Cl at 50 °C for 1 h furnished the desired
cis-2,6-disubstituted tetrahydropyran
14a in 56% yield. The yield of
cis-2,6-disubstituted tetrahydropyran
14a in this reaction was exactly the same as the yield from bis-propargyl alcohol
13a (
Table 1, entry 5). This result most likely indicates that the reaction proceeded from bis-propargylic alcohol
13a through the tetrahydropyran
14′a as the intermediate to
cis-2,6-disubstituted tetrahydropyran
14a. (
Scheme 5).
In the study, as shown in
Table 1, we confirmed the termination of the reaction by the disappearance of bis-propargylic alcohol
13a. However, from the studies in the previous section (
Scheme 4 and
Scheme 5), it was estimated that tetrahydropyran
14′a was likely to be an intermediate in this reaction. Therefore, it was decided to change the confirmation of the end of the reaction by the disappearance of tetrahydropyran
14′a and to examine the reaction again (
Table 2).
Entries 1 and 2 in
Table 2 are the results shown in
Table 1, entries 4 and 5. The reaction of entry 2 (reaction time: 2 h) was extended until the disappearance of intermediate
14′a was confirmed, resulting in an extension of the reaction time to 5 h and a slightly higher yield of 61%. (entry 3). When the reaction was carried out with the reduction of the catalytic amount to 5 mol% Ph
3PAuCl and 5 mol% AgNTf
2, the yield of the product
14a was slightly lower, 48% (entry 4). Furthermore, when the reaction with 10 mol% Ph
3PAuCl and 10 mol% AgNTf
2 was conducted with reducing the amount of water to 1 eq., the tetrahydropyran
13a did not disappear even after 24 h, and the yield of the desired
cis-2,6-disubstituted tetrahydropyran
14a was obtained in 41% yield (entry 5). This result indicates that additional water is essential for the reaction to proceed efficiently. On the other hand, the reaction was conducted only with the addition of water (10 eq.) without methanol, resulting in a low yield of the desired
cis-2,6-disubstituted tetrahydropyran
14a (entry 6). Finally, the optimal reaction conditions for preparation of the desired
cis-2,6-disubstituted tetrahydropyran
14a from bis-propargylic alcohol
13a were found to be Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of MeOH (2 eq.) and H
2O (10 eq.) in ClCH
2CH
2Cl stirred at 50 °C for 5 h.
Next, we examined the scope of the reaction with bis-propargylic alcohols
13 bearing other substituents of the alkyne moiety (
Table 3). Treatment of bis-propargylic alcohols
13b bearing a hexyl group at the alkyne terminus with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of MeOH (2 eq.) and H
2O (10 eq.) at 50 °C in 1,2-dichloroethane for 3.5 h afforded the corresponding
cis-2,6-disubstituted tetrahydropyran
14b in 59% (entry 2). The reaction with bis-propargylic alcohol
13c having
n-Hex and Ph groups as substituents at the alkyne terminus also furnished the corresponding
cis-2,6-disubstituted tetrahydropyran
14c in 61% yield (entry 3).
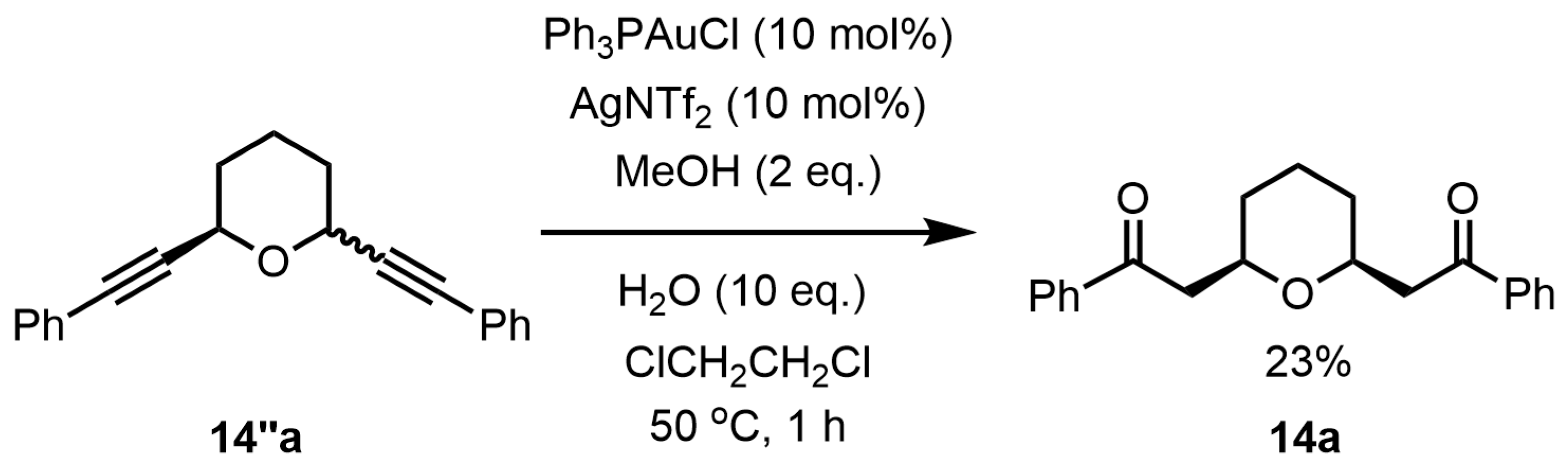
Next, the transformation of the triple bond in tetrahydropyran
14 to the carbonylmethyl group via hydration reaction was investigated. Treatment of tetrahydropyran
14″a with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of MeOH (2 eq.) and H
2O (10 eq.) at 50 °C in 1,2-dichloroethane for 1 h afforded
cis-2,6-disubstitued tetrahydropyran
14a in a 23% yield (
Scheme 6).
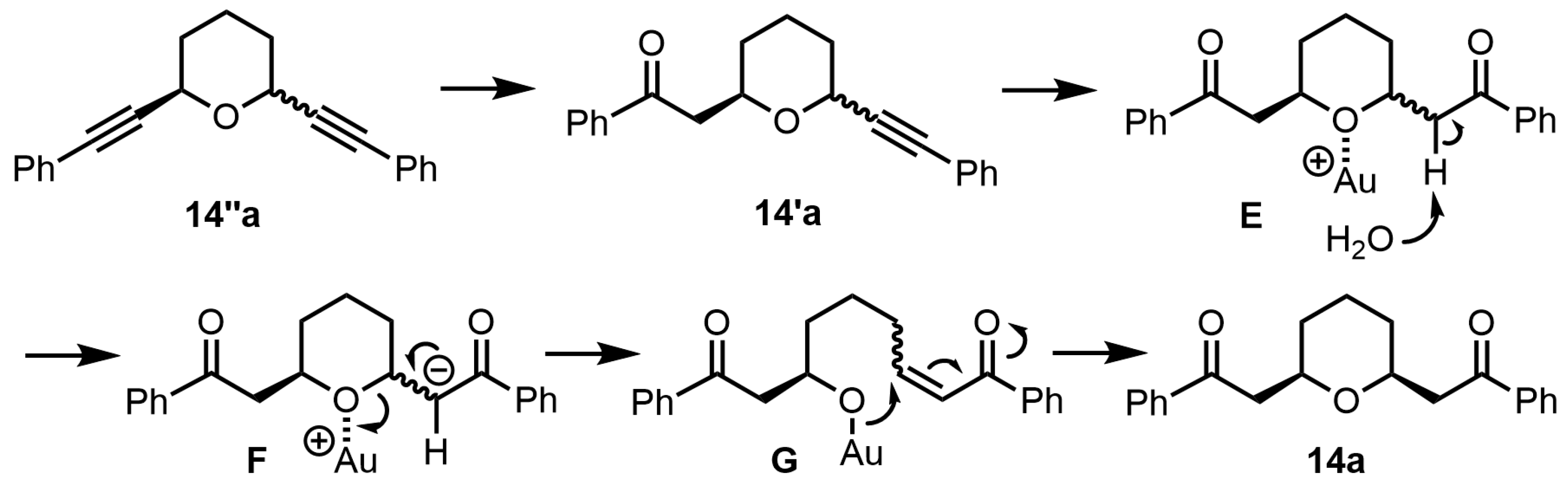
If the hydration reaction occurred without the ring-opening reaction of the tetrahydropyran ring, the product, tetrahydropyran 14a, should be a mixture of cis- and trans-forms. In practice, however, only cis-2,6-disubstituted tetrahydropyran 14a was obtained as a product in the reaction, so it is assumed that the ring-opening reaction occurred during the hydration reaction.
The plausible reaction mechanism for the preparation of the
cis-2,6-disubstituted tetrahydropyran
14a from tetrahydropyran
14″a is shown in
Scheme 7. First, the first hydration reaction occurs at one triple bond in tetrahydropyran
14″a, forming the mixture of
cis- and
trans-2,6-disubstituted tetrahydropyran
14′a bearing a carbonylmethyl group (
14″a→
14′a). Next, the second hydration reaction occurs at the other triple bond in tetrahydropyran
14′a to form the mixture of
cis- and
trans-2,6-disubstituted tetrahydropyran
14a. Then, the coordination of the gold catalyst with the oxygen atom of tetrahydropyran
14a (
E) and the water-induced elimination of α-hydrogen result in a ring-opening reaction by reverse oxa-Michael addition (
F→
G) and a ring-closing reaction by oxa-Michael addition (
G→
14a), ultimately yielding stable
cis-2,6-disubstituted tetrahydropyran
14a.
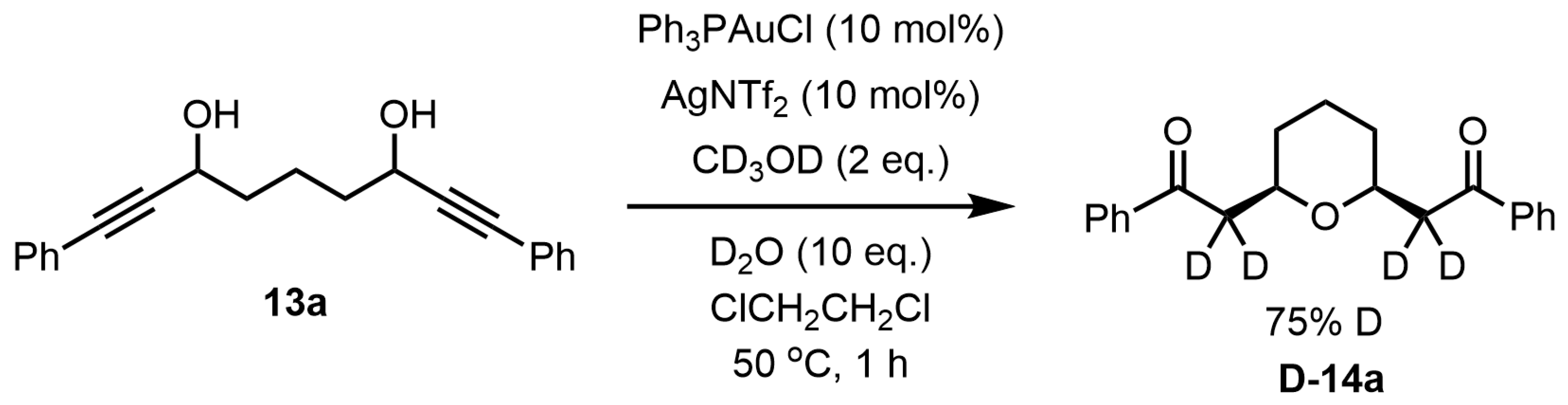
Deuteration experiments were conducted to understand the reaction mechanism. Treatment of bis-propargylic alcohol
13a with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of CD
3OD (2 eq.) and D
2O (10 eq.) at 50 °C in 1,2-dichloroethane for 1 h afforded the desired 2,6-
cis-disubstituted tetrahydropyran
D-14a, showing that the methylene groups at 2,6-positions were both 75% deuterated in the
1H NMR spectrum (
Scheme 8).
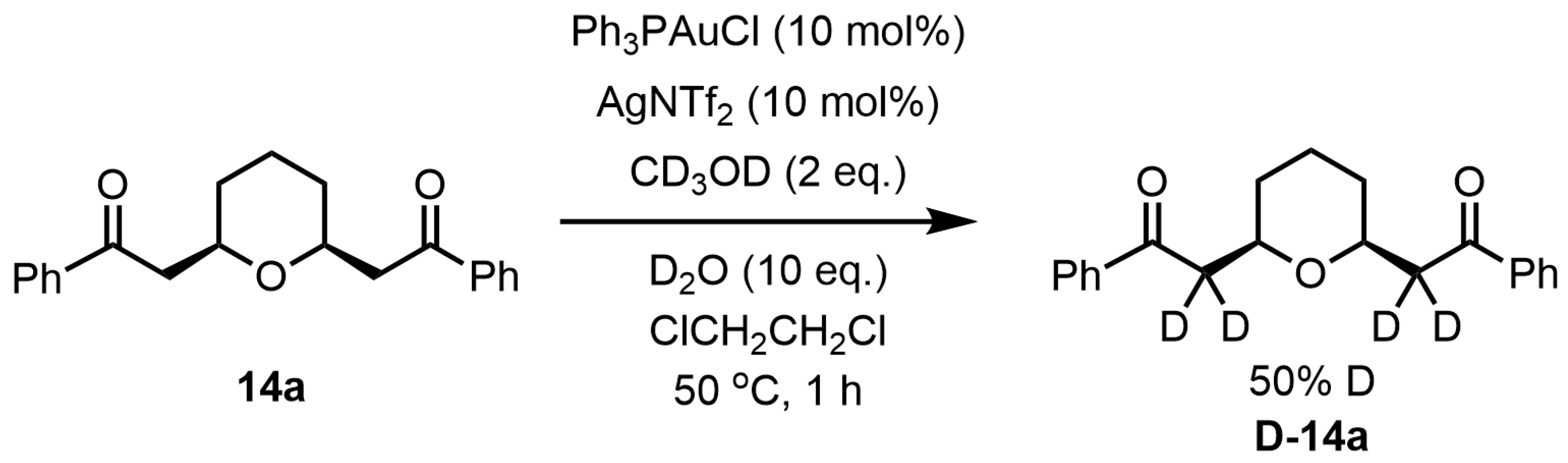
Next, deuteration experiments were performed on
cis-2,6-disubstituted tetrahydropyran
14a. Treatment of the tetrahydropyran
14a with Ph
3PAuCl (10 mol%) and AgNTf
2 (10 mol%) in the presence of CD
3OD (2 eq.) and D
2O (10 eq.) at 50 °C for 1 h in 1,2-dichloroethane afforded the tetrahydropyran
D-14a, showing that the methylene groups at 2,6-positions were both 50% deuterated in the
1H NMR spectrum (
Scheme 9). Although the result of this deuteration experiment indicates that deuteration also occurs after the formation of tetrahydropyran
14a, the deuteration rate is higher in the reaction from bis-propargyl alcohol
13a (
Scheme 8) than in the reaction from tetrahydropyran
14a (
Scheme 9), which suggests that MeOH and H
2O are involved in the formation of tetrahydropyran
14a from bis-propargyl alcohol
13a.
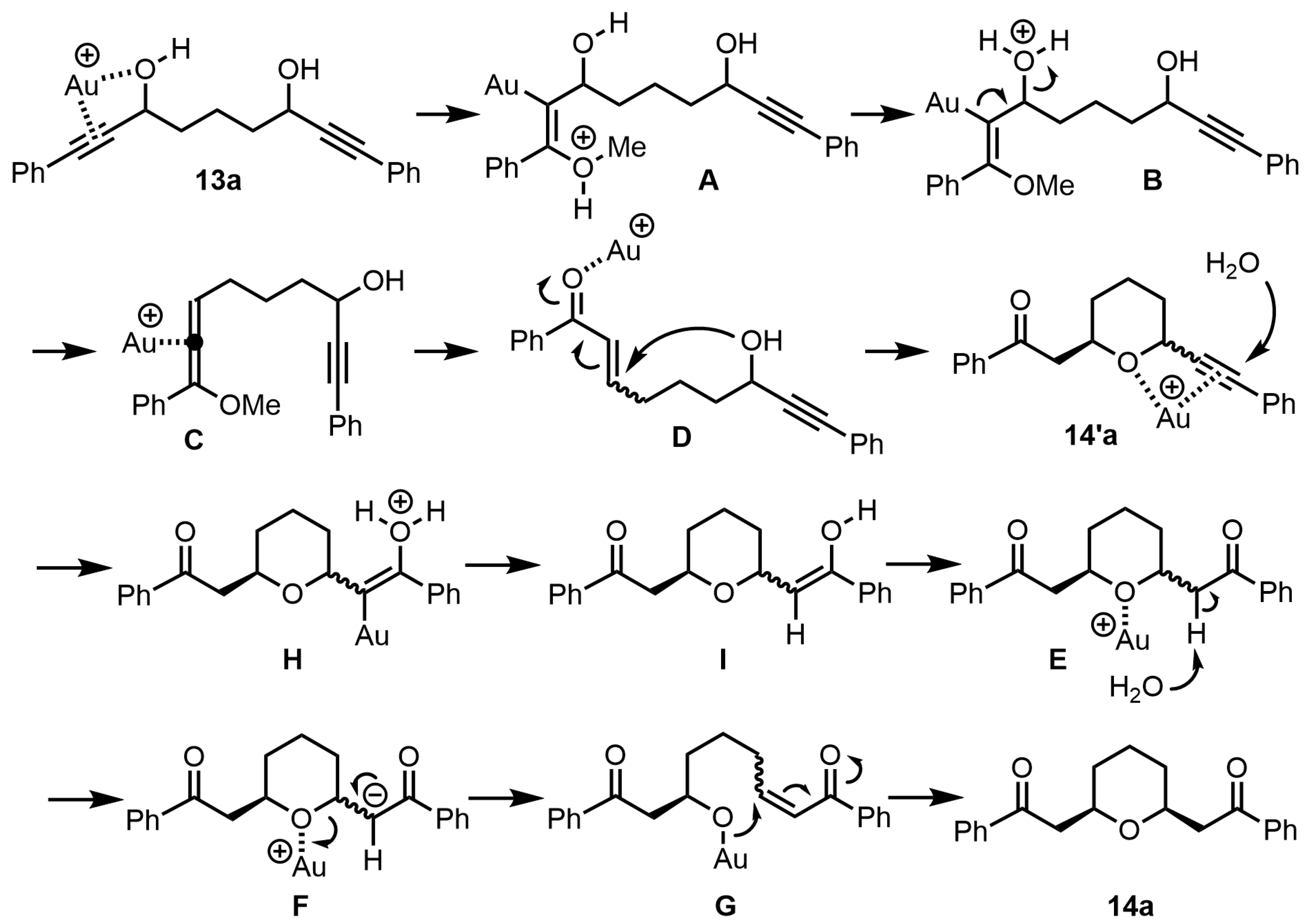
The plausible reaction mechanism for the preparation of
cis-2,6-disubstituted tetrahydropyran
14a from bis-propargyl alcohol
13a is shown in
Scheme 10. First, the coordination of the gold (I) catalyst to the triple bond and hydroxyl group of bis-propargylic alcohol
13a results in a nucleophilic attack by MeOH on the activated triple bond to form vinyl gold species
A (
13a→
A). Next, the carbon-gold bond in vinyl gold species
A is cleaved with the elimination of water, forming the allene intermediate
C (
A→
B→
C). Subsequently, the addition of water transforms the allene intermediate
C to α,β-enone
D, completing the Meyer–Schuster rearrangement reaction (
C→
D). Furthermore, the gold catalyst coordinates with the carbonyl oxygen to enhance the reactivity of α,β-enone, resulting in the intramolecular oxa-Michael addition reaction to afford intermediate
14′a (
D→
14′a). Then, the gold (I) catalyst coordinates with the other triple bond and the oxygen atom of tetrahydropyran
14′a to bring about the hydration reaction, giving the mixture of
cis- and
trans-2,6-disubstituted tetrahydropyran
14a (
H→
I→
E). The reaction mechanism for the formation of
cis-2,6-disubstituted tetrahydropyran
14a from the mixture of
cis- and
trans-2,6-disubstituted tetrahydropyran is described in
Scheme 7.
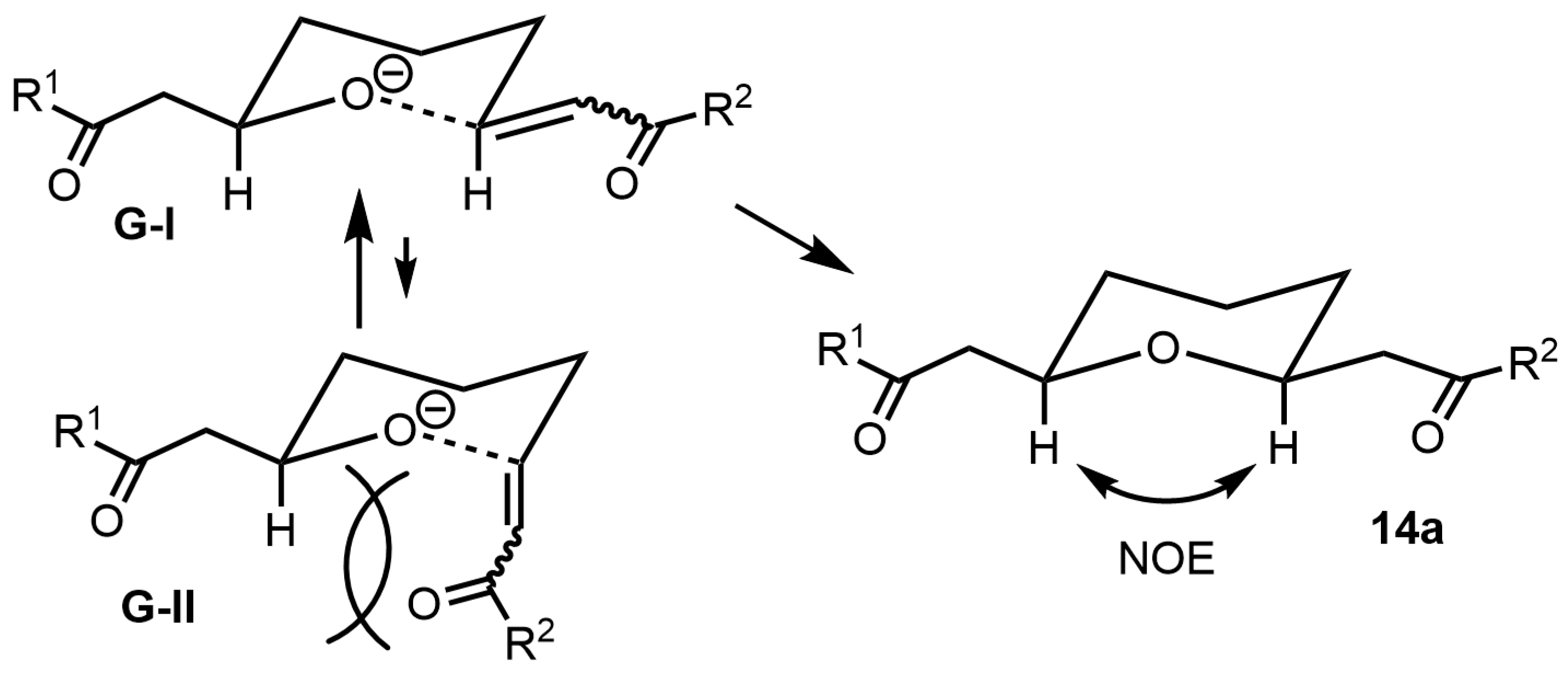
The stereochemistry of
cis-2,6-disubstituted tetrahydropyran
14c was confirmed by NOE measurements (
Figure 2). The stereochemical outcome of the reaction can be explained based on transition-state structures
G-I and
G-II (
Figure 2). During the equilibration between the reverse oxa-Michael addition and oxa-Michael addition (
Scheme 10), the bulky substituent (α,β-enone) occupies a pseudo-equatorial position to avoid 1,3-diaxial interactions (
G-II). As the α,β-enone group is bulkier than hydrogen, it tends to take a pseudo-equatorial position (
G-I) rather than a pseudoaxial position (
G-II) to avoid 1,3-diaxial interaction, resulting in stereoselective synthesis of
cis-2,6-disubstituted tetrahydropyran
14 [
13,
27].
3. Materials and Methods
3.1. General Information
1H and 13C NMR spectra were recorded with a JEOL JNM-AL300 (Japan Electron Optics Laboratory Co., Ltd., Tokyo, Japan) or BRUKER AV-300 spectrometer (Bruker, Billerica, MA, USA) at room temperature, with tetramethylsilane as an internal standard (CDCl3 solution). Chemical shifts were recorded in ppm and coupling constants (J) in Hz. Infrared (IR) spectra were recorded with a Shimadzu FTIR-8200A spectrometer (Shimadzu Corporation, Kyoto, Japan). Mass spectra were recorded on JEOL JMS-700 spectrometers (Japan Electron Optics Laboratory Co., Ltd., Tokyo, Japan). Merck silica gel 60 (1.09385) (Merck, Darmstadt, Germany) and Merck silica gel 60 F254 (Merck, Darmstadt, Germany) were used for column chromatography and thin-layer chromatography (TLC), respectively.
1,9-Diphenylnona-1,8-diyne-3,7-diol (13a): Colorless oil, IR (KBr) 3319, 3055, 2947, 2864, 2228, 1599, 1489, 1443, 1026, 756, 691 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.42–7.40 (4H, m), 7.31–7.26 (6H m), 4.65 (2H, t, J = 6.0 Hz), 2.07 (2H, br s), 1.90–1.79 (6H, m); 13C-NMR (75 MHz, CDCl3) δ 131.7, 128.4, 128.3, 122.5, 89.8, 85.1, 62.8, 37.3, 21.0; HRMS (EI) m/z calcd for C21H20O2 [M]+ 304.1463, found 304.1452.
Henicosa-7,14-diyne-9,13-diol (13b): Colorless oil, IR (KBr) 3362, 2928, 2856, 2233, 1464, 1082, 1022 cm−1; 1H-NMR (300 MHz, CDCl3) δ 4.37 (4H, br s), 2.20 (2H, td, J = 7.1, 1.8 Hz), 1.74–1.68 (3H, m), 1.65–1.59 (5H, m), 1.53–1.48 (3H, m), 1.40–1.27 (13H, m), 0.89 (6H, t, J = 6.6 Hz); 13C-NMR (75 MHz, CDCl3) δ 85.6, 81.1, 62.4, 37.7, 31.3, 28.6, 28.5, 22.5, 21.0, 18.6, 14.0; HRMS (FAB) m/z calcd for C21H37O2 [M + H]+ 321.2794, found 321.2740.
1-Phenylpentadeca-1,8-diyne-3,7-diol (13c): Colorless oil, IR (KBr) 3354, 3082, 2932, 2860, 2233, 1599, 1490, 1026 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.45–7.41 (2H, m), 7.32–7.26 (3H, m), 4.62 (1H, t, J = 6.0 Hz), 4.40–4.38 (1H, m), 2.19 (2H, td, J = 7.0, 0.9 Hz), 2.07 (1H, br s), 1.88–1.82 (3H, m), 1.79–1.69 (4H, m), 1.53–1.44 (2H, m), 1.38–1.27 (6H, m), 0.88 (3H, t, J = 7.5 Hz); 13C-NMR (75 MHz, CDCl3) δ 131.6, 128.23, 128.16, 122.6, 90.0, 85.6, 84.8, 81.0, 62.6, 62.4, 37.5, 37.3, 31.2, 28.6, 28.5, 22.4, 20.9, 18.6, 14.0; HRMS (EI) m/z calcd for C21H28O2 [M]+ 312.2089, found 312.2076.
3.2. Synthetic Procedure of 2,6-Disubstituted Tetrahydropyran 14′a from Bis-Propargylic Alcohol 13a
MeOH (13 μL, 0.33 mmol, 2 eq.), Ph3PAuCl (8.1 mg, 0.016 mmol, 10 mol%) and AgNTf2 (6.4 mg, 0.016 mmol, 10 mol%) were added to a solution of bis-propargylic alcohol 13a (50 mg, 0.16 mmol) in toluene (5 mL) at room temperature, and the mixture was stirred at reflux for 3.5 h. The solvent was removed in vacuo and the crude product was subjected to SiO2 column chromatography (hexane:CH2Cl2 = 2:1) to give the mixture of 2,6-disubstituted tetrahydropyran cis-14′a (17 mg, 34%) and trans-14a′ (20 mg, 40%).
Phenyl-2-[(2R*,6S*)-6-(phenylethynyl)tetrahydro-2H-pyran-2-yl]ethan-1-one (cis-14′a). Colorless oil (hexane:CH2Cl2 = 2:1), IR (KBr) 3059, 2926, 2855, 2230, 1684, 1597, 1491, 1448, 1047 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.97 (2H, d, J = 7.2 Hz), 7.57 (1H, t, J = 7.4 Hz), 7.49–7.42 (4H, m), 7.30–7.27 (3H, m), 4.43 (1H, dd, J = 11.0, 2.1 Hz), 4.13–4.06 (1H, m), 3.45 (1H, dd, J = 16.8, 5.1 Hz), 3.07 (1H, dd, J = 16.8, 7.2 Hz), 1.96–1.75 (4H, m), 1.42–1.29 (2H, m); 13C-NMR (75 MHz, CDCl3) δ 137.1, 133.2, 131.9, 128.6, 128.3, 128.2, 128.1, 122.6, 88.4, 84.4, 74.6, 68.9, 45.3, 32.5, 30.9, 23.2; HRMS (FAB) m/z calcd for C21H21O2 [M + H]+ 305.1542, found 305.1538.
1-Phenyl-2-[(2R*,6R*)-6-(phenylethynyl)tetrahydro-2H-pyran-2-yl]ethan-1-one (trans-14′a). Colorless oil (hexane:CH2Cl2 = 2:1), IR (KBr) 3061, 2926, 2853, 1686, 1597, 1489, 1448, 1038 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.97 (2H, d, J = 7.5 Hz), 7.56 (1H, t, J = 7.5 Hz), 7.48–7.41 (4H, m), 7.31–7.29 (3H, m), 4.96 (1H, d, J = 4.2 Hz), 4.67–4.59 (1H, m), 3.28 (1H, dd, J = 15.6, 6.0 Hz), 3.03 (1H, dd, J = 15.6, 6.6 Hz), 2.02 (1H, tt, J = 12.4, 3.6 Hz), 1.88–1.75 (4H, m), 1.43–1.30 (2H, m); 13C-NMR (75 MHz, CDCl3) δ 198.1, 137.2, 133.0, 131.8, 128.5, 128.2, 122.8, 87.4, 86.8, 68.8, 65.9, 45,4, 31.6, 30.4, 29.7, 19.4; HRMS (FAB) m/z calcd for C21H21O2 [M + H]+ 305.1542, found 305.1536.
3.3. General Procedure for Gold-Catalyzed Synthesis of 2,6-Disubstituted Tetrahydropyrans 14a from Bis-Propargylic Alcohols 13
Ph3PAuCl (10 mol%) and AgNTf2 (10 mol%) were added to a solution of bis-propargylic alcohol 13, MeOH (2 eq.) and H2O (10 eq.) in ClCH2CH2Cl at room temperature, and the mixture was stirred at 50 °C. After complete consumption of 2,6-disubstituted tetrahydropyran 14′ (the reaction was monitored by thin layer chromatography; usually within 5 h), the solvent was removed in vacuo and the crude product was subjected to SiO2 column chromatography (eluents = hexane:CH2Cl2) to give cis-2,6-disubstituted tetrahydropyran 14.
2,2’-[(2R*,6S*)-Tetrahydro-2H-pyran-2,6-diyl]bis(1-phenylethan-1-one) (cis-14a). Bis-propargylic alcohol 13a (30 mg, 0.099 mmol), MeOH (8.0 μL, 0.20 mmol, 2 eq.), H2O (18 μL, 0.99 mmol, 10 eq.), Ph3PAuCl (4.9 mg, 0.0099 mmol, 10 mol%) and AgNTf2 (3.9 mg, 0.0099 mmol, 10 mol%) in ClCH2CH2Cl (5 mL) furnished cis-14a (20 mg, 61%) as a colorless oil (hexane:CH2Cl2 = 6:1).
IR (KBr) 3063, 2926, 2855, 1684, 1597, 1510, 1448, 1063 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.92–7.89 (4H, m), 7.53 (2H, tt, J = 7.2, 1.5 Hz), 7.45–7.39 (4H, m), 4.05–3.97 (2H, m), 3.24 (2H, dd, J = 15.9, 6.3 Hz), 2.94 (2H, dd, J = 15.9, 6.6 Hz), 1.86–1.74 (2H, m), 1.68–1.57 (2H, m), 1.35–1.27 (2H, m); 13C-NMR (75 MHz, CDCl3) δ 198.3, 137.3, 133.0, 128.5, 128.2, 74.7, 45.4, 31.4, 23.3; HRMS (EI) m/z calcd for C21H22O3 [M]+ 304.1569, found 322.1570.
The
1H-NMR and
13C-NMR data are identical with reported values [
13].
1,1’-[(2R*,6S*)-Tetrahydro-2H-pyran-2,6-diyl]bis(octan-2-one) (cis-14b). Bis-propargylic alcohol 13b (30 mg, 0.094 mmol), MeOH (7.6 μL, 0.19 mmol, 2 eq.), H2O (17 μL, 0.94 mmol, 10 eq.), Ph3PAuCl (4.6 mg, 0.0094 mmol, 10 mol%) and AgNTf2 (3.6 mg, 0.0094 mmol, 10 mol%) in ClCH2CH2Cl (5 mL) furnished cis-14b (19 mg, 59%) as a colorless oil (hexane:CH2Cl2 = 3:1) as a colorless oil.
IR (KBr) 2930, 2858, 1715, 1373, 1080 cm−1; 1H-NMR (300 MHz, CDCl3) δ 3.84–3.76 (2H, m), 2.59 (2H, dd, J = 15.2, 8.1 Hz), 2.43–2.33 (6H, m), 1.84–1.79 (1H, m), 1.68–1.50 (8H, m), 1.27–1.15 (13H, m), 0.88 (6H, t, J = 6.6 Hz); 13C-NMR (75 MHz, CDCl3) δ 209.5, 74.4, 49.4, 43.6, 31.6, 31.1, 28.8, 23.5, 23.2, 22.5, 14.0; HRMS (EI) m/z calcd for C21H38O3 [M]+ 338.2821, found 338.2812.
1-[(2R*,6S*)-6-(2-Oxo-2-phenylethyl)tetrahydro-2H-pyran-2-yl]octan-2-one (cis-14c). Bis-propargylic alcohol 13c (30 mg, 0.096 mmol), MeOH (7.8 μL, 0.19 mmol, 2 eq.), H2O (18 μL, 0.94 mmol, 10 eq.), Ph3PAuCl (4.8 mg, 0.0096 mmol, 10 mol%) and AgNTf2 (3.8 mg, 0.0096 mmol, 10 mol%) in ClCH2CH2Cl (5 mL) furnished cis-14c (19 mg, 59%) as a colorless oil (hexane:CH2Cl2 = 2:1) as a colorless oil.
IR (KBr) 3069, 2932, 2860, 1713, 1686, 1597, 1491, 1448 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.97–7.92 (2H, m), 7.56 (1H, tt, J = 7.5, 1.5 Hz), 7.49–7.42 (2H, m), 4.04–3.94 (1H, m), 3.86–3.77 (1H, m), 3.26 (1H, dd, J = 15.6, 6.6 Hz), 2.89 (1H, dd, J = 15.6, 6.0 Hz), 2.58 (1H, dd, J = 15.3, 7.8 Hz), 2.40–2.30 (3H, m), 1.89–1.60 (4H, m), 1,51–1.40 (2H, m), 1.29–1.18 (8H, m), 0.86 (3H, t, J = 6.6 Hz); 13C-NMR (75 MHz, CDCl3) δ 209.7, 198.4, 137.3, 133.0, 128.5, 128.2, 74.7, 74.6, 49.5, 45.2, 43.5, 31.6, 31.2, 28.8, 23.4, 23.2, 22.5, 14.0; HRMS (EI) m/z calcd for C21H30O3 [M]+ 330.2195, found 330.2202.
3.4. Synthetic Procedure of 2,6-Disubstituted Tetrahydropyran 14″a from Bis-Propargylic Alcohols 13a
AuBr3 (4.3 mg, 0.00099 mmol, 10 mol%) were added to a solution of bis-propargylic alcohol 13a (30 mg, 0.099 mmol) in ClCH2CH2Cl (5 mL) at room temperature, and the mixture was stirred at reflux for 48 h. The solvent was removed in vacuo and the crude product was subjected to SiO2 column chromatography (hexane:CH2Cl2) to give the mixture of 2,6-disubstituted tetrahydropyran cis-14″a (7.4 mg, 26% yield) and trans-14″a (5.6 mg, 20% yield).
(2R*,6S*)-2,6-Bis(phenylethynyl)tetrahydro-2H-pyran (cis-14″a). Colorless oil (hexane:CH2Cl2 = 5:2), IR (KBr) 3080, 2947, 2922, 2850, 2360, 1599, 1491, 1379, 1072 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.46–7.42 (4H, m), 7.32–7.28 (6H, m), 4.43 (2H, dd, J = 10.8, 2.4 Hz), 2.00–1.91 (3H, m), 1.87–1.78 (2H, m), 1.72–1.61 (1H, m); 13C-NMR (75 MHz, CDCl3) δ 131.8, 128.4, 128.2, 122.6, 87.8, 84.9, 68.9, 32.0, 29.7, 23.3; HRMS (EI) m/z calcd for C21H18O [M]+ 286.1358, found 286.1361.
(2R*,6R*)-2,6-Bis(phenylethynyl)tetrahydro-2H-pyran (trans-14′a). Colorless oil (hexane:CH2Cl2 = 2:1), IR (NaCl) 3055, 2924, 2851, 2359, 2235, 1599, 1491, 1194, 1026 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.49–7.45 (4H, m), 7.33–7.30 (6H, m), 5.05–5.01 (2H, m), 2.00–1.90 (4H, m), 1.84–1.77 (2H, m); 13C-NMR (75 MHz, CDCl3) δ 131.8, 128.4, 128.2, 122.6, 87.5, 86.0, 64.6, 31.2, 29.7, 19.3; HRMS (EI) m/z calcd for C21H18O [M]+ 286.1358, found 286.1360.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


